












HTML
-
Bacteriophages predominate in the biosphere and outnumber their hosts by at least one order of magnitude (Srinivasiah et al., 2008). They have been used for over 90 years as an alternative to antibiotics in Eastern Europe (Deresinski, 2009). With the increasing emergence of antibiotic resistance, the therapeutic potential of bacteriophages is being reevaluated (Kutter et al., 2010). Until recently, phages possessing dsDNA genomes have been the most thoroughly studied. There is a great deal of data available on their evolution, diversity, and host relationships (Krupovic et al., 2011). However, technological advances in ssDNA amplification and sequencing have shown that viruses with ssDNA genomes are more prevalent than previously recognized, and it has recently been discovered that these viruses are important members of the biosphere (Desnues et al., 2008). One of the most commonly retrieved ssDNA viruses in the environment belongs to the family Microviridae.
Members of the family Microviridae are non-enveloped, polyhedral, and icosahedral lytic viruses, consisting of ssDNA circular genomes (Roux et al., 2012). Microviridae-like sequences have been found in multiple ecosystems, including human stool and coral samples (Hopkins et al., 2014). Microviridae are further divided into four sub-groups based on genomic and structural characteristics that include microviruses (genus Microvirus), gokushoviruses (subfamily Gokushovirinae), alphaviruses (subfamily Alphavirinae), and recently described pichoviruses (subfamily Pichovirinae) (Roux et al., 2012).
In the present study, we reported the full genome sequence of a novel bacteriophage, designated as IME-16, via screening pathogens from a throat swab obtained from a patient with an unexplained fever. The viral DNA and RNA were extracted using a High Pure Viral RNA Kit (Roche, Germany). Reverse transcription was performed using a SuperScript Ⅲ First-Str and kit (Invitrogen, USA). DNA was amplified using a non-specific multiple displacement amplification (MDA) method (New Engl and Biolabs, UK) (Willner et al., 2009). Purified amplification products were sequenced using a high-throughput sequencing (HTS) platform IonTorrent PGM Sequencer (Thermo Fisher Scientific, USA).
After sequencing, a total of 2, 831, 214 raw reads were obtained. Low quality and low complexity sequences were removed using NGS QC Toolkit v2.3.3 (Patel and Jain, 2012). After data filtering, 1, 179, 588 clean reads with a mean length of 183 bp, totaling 538 Mb, were obtained. The clean reads were first searched against the human genome (hg19) database using BLAT. Then, the 203 Mb that did not map to any sequences in the hg19 database were searched against the NCBI NT database using BLASTn (-v 3 -b 5 -W 30 -e 1e-5). A total of 46 Mb of the data matched sequences in the NT database. The remaining 160 Mb of data, containing 574, 032 sequences, were extracted and assembled using Roche Newbler version 2.5 (Roche Applied Science). A contig of 5, 775 bp that could be circularized was generated. A total of 2, 368 reads that did not match the NT database provided 99 coverage of the contig. The maximum coverage was 142×. The contig was then used to search against the NCBI NR database using BLASTx (default), which showed that it displayed homology with members of the family Microviridae. Thus, the 5, 755-bp sequence was proposed to be a c and idate bacteriophage genome belonging to a member of the Microviridae family; this bacteriophage was designated as IME-16.
The circular ssDNA of IME-16 possessed an overall G+C content of 37.1% and A, C, G, and T frequencies of 27.4%, 19.5%, 17.5%, and 35.6%, respectively. The genome size of IME-16 is larger than that of gokushoviruses and smaller than that of microviruses. Genome annotations and open reading frames (ORFs) were completed using the RAST server (Aziz et al., 2008) and Glimmer v3.02 (Delcher et al., 2007). Seven ORFs were identified in the IME-16 genome, which had lengths of 120-2121 bp. Five out of the seven ORFs presented ATG as a start codon and were preceded by a ribosome-binding site. ORFs were further analyzed for protein sequence homology by using BLASTp (default) programs. The seven ORFs encoded putative proteins with lengths of 40-707 amino acids (aa) (Table 1). The predicted ORFs covered 5, 163 bp, resulting in a coding density of 89.4%. A map of the predicted ORFs was then generated using CGView (Stothard and Wishart, 2005), with gene features as shown in Figure 1A. The prediction of phage-encoded tRNA genes was performed by using tRNAscan-SE v. 1.21 (Schattner et al., 2005). The absence of any tRNA-encoding sequences suggested that the phage may have adapted ideally to its host organism and requires no additional tRNAs of its own. Initial homology analysis using BLASTn and BLASTp showed no significant hits. We therefore, performed a homology search analysis against additional genome sequences of Microviridae from previously reported metagenomic studies (Roux et al., 2012) using MatGAT v. 2.02 (Campanella et al., 2003). A homology search and analysis of similarity indices showed that amino acid sequences encoded by ORF2 and ORF7 showed 27% and 22% similarity to the genomes of the alphaviruses, a sub-family in Microviridae. Based on this sequence similarity and the overall genome organization analysis, IME-16 was predicted to be most closely related to members of the genus Alphavirinae. The peptide sequences encoded by the remaining putative ORFs showed no satisfactory similarity to amino acid sequences of the currently available phage proteins.
ORF Start Stop Nucleotide Size (aa) Start codon Function 1 22 1218 1197 399 ATG M16p1 2 1463 2032 570 190 ATG Replication initiator protein 3 2214 2819 606 202 TTG M16p2 4 2876 3088 213 71 ATG M16p3 5 3152 3271 120 40 TTG M16p4 6 3261 3596 336 112 ATG M16p5 7 3609 5729 2121 707 ATG Major capsid protein Table 1. Summary and functional categories of the predicted ORFs of IME-16.
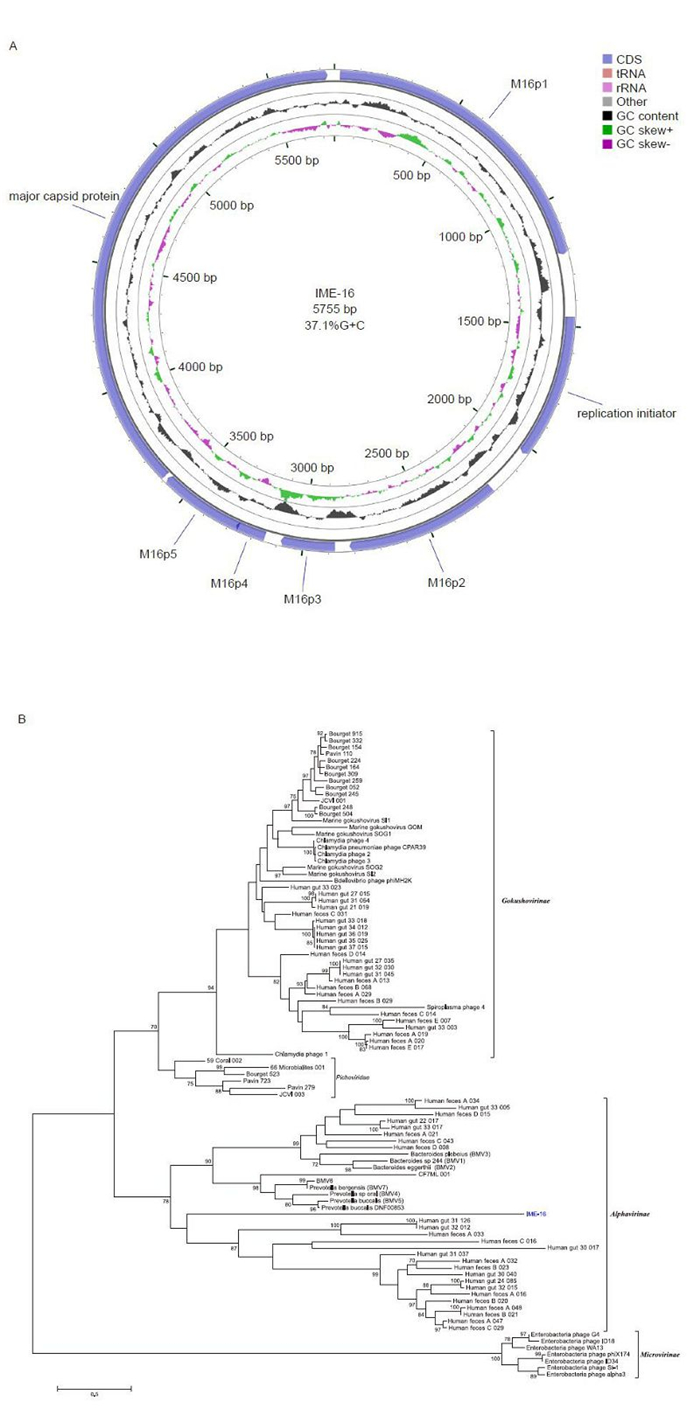
Figure 1. Genomic analysis of IME-16. (A) Genetic and physical map of IME-16. The circle with green and violet peaks represents the GC skew and the circle with black peaks shows the G+C content. (B) Phylogenetic analysis of the IME-16 major capsid protein. Tree constructed by the maximum-likelihood method. The values at the nodes indicate the bootstrap scores using 1, 000 replicates.
ORF2 was predicted to encode a protein of 190 aa with a maximum similarity: identity ratio of 32.4:18.3 to the replication initiator protein (RIP) of Pavin_723 from Pichovirinae. In order to further confirm the presence of functional RIP in IME-16, we constructed multiple sequence alignments of selected RIP from different phages and determined the presence of regions known to be specific to RIPs in Microviridae. Three conserved motifs, Motif Ⅰ: 52-LLTLTY-57, Motif Ⅱ: 113-KRPHYHLLG-121, and Motif Ⅲ: 165-GAAYASKYV-173 (Ilyina and Koonin, 1992) characteristic to rolling circle replication proteins were found in IME-16 ORF2 (Supplementary Figure S1). Furthermore, two conserved tyrosine (Y) residues previously designated as a signature of superfamily Ⅰ RCR proteins were also found in motif Ⅲ at position 168 and 172 of IME-16 ORF2, further confirming that ORF2 of IME-16 encodes a functional RIP. ORF7 was predicted to encode a protein of 707 aa with similarity: identity ratios of 38.1:18.8 and 36.3:18.9 to the major capsid protein (MCP) of CF7ML phage closely followed by Prevotella buccalis (BMV5), both from the Alphavirinae group.
To better underst and the relationship between IME-16 and other members of the Microviridae family, a phylogenetic analysis was performed. Ninety-seven major capsid protein (MCP) sequences of phages from the Microviridae family (Roux et al., 2012), including that of IME-16, were aligned using MUSCLE within the Molecular Evolutionary Genetic Analysis (MEGA) (version 6.0.5) software suite (Tamura et al., 2013). A phylogenetic tree was constructed using the maximum likelihood (ML) method and the robustness of trees was assessed by bootstrap analysis of 1000 replicates. Four well-characterized clades (bootstrap values > 75) were formed, representing the genus Microvirus and subfamilies Gokushovirinae, Alphavirinae, and Pichovirinae (Figure 1B). In agreement with the similarity index analysis of MCPs, the IME-16 MCP grouped along with members of Alphavirinae, indicating that IME-16 is likely to be more closely related to the members of the Alphavirinae than to other sub-families. It is also interesting to note that, despite grouping within the Alphavirinae cluster, the clustering appears to be somewhat distinct, suggesting that IME-16 itself is a distinct member of the Alphavirinae group or might represent a new group within Microviridae. However, this can only be confirmed once additional sequencing data becomes available.
Nucleotide sequence accession number: The fulllength genome sequence of IME-16 was deposited into the GenBank database and assigned the accession number KM276541.
-
This research was supported by a grant from the National Hi-Tech Research and Development (863) Program of China (No. 2012AA022003 and No.2014AA021402), the China Mega-Project on Infectious Disease Prevention (No. 2013ZX10004605, No. 2011ZX10004001, No. 2013ZX10004607-004 and No. 2013ZX10004217-002-003) and the State Key Laboratory of Pathogen and BioSecurity Program (No. SKLPBS1113). The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
Supplementary Figure S1 is available on the website of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-
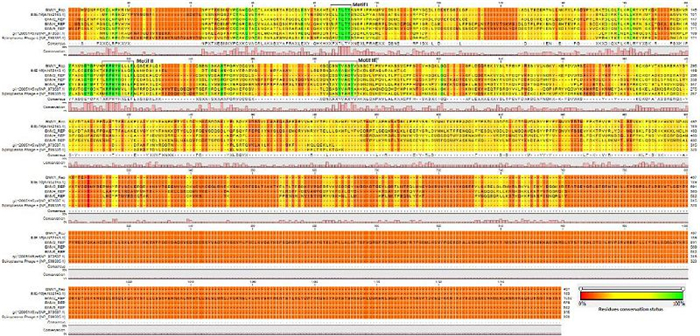
Figure S1. Sequence analysis of the replication initiator protein of IME-16. Alignment of the three conserved motifs (Ⅰ-Ⅲ) of superfamily Ⅰ rolling circle replication proteins with corresponding motifs from the putative replication proteins of viruses from micro-, alpha-, gokusho-and pichoviruses.

 DownLoad:
DownLoad: