










HTML
-
Antibiotics are widely used to control bacterial diseases but it is very easy for the pathogens to become resistant to antibiotics(Dupont et al., 2007). Currently, the threat of multi-resistant bacteria is increasing, and antibiotic therapy is becoming more and more ineffective, so it is important to search for alternative antimicrobials(Oliveira et al., 2014).
Endolysins are encoded by bacteriophages and released at the terminal stage of the replication cycle to degrade the peptidoglycan bacterial wall from within(Oliveira et al., 2013). They show promise in being used as antibacterials for their high specificity, nontoxicity, high efficiency and lack of drug resistance(Nelson et al., 2012; Trudil, 2015). However, endolysins have been reported to destroy mainly Gram-positive organisms(such as Staphylococcus, Streptococcus and Listeria)but not Gram-negative organisms(such as Salmonella, Escherichia coli, and Pseudomonas aeruginosa), owing to their inability to cross the bacterial outer membrane(Fischetti, 2010). Interestingly, recent studies have revealed that several phage endolysins may be effective against Gram-negative bacteria(Lim et al., 2014; Endersen et al., 2015).
Certain permeabilizers that may increase the access of endolysins to the Gram-negative peptidoglycan layer have been reported. EDTA is a well-known outer membrane permeabilizer that enhances the activity of these bacterial cell wall hydrolases by removing divalent cations from their binding sites and causing outer membrane disruption(Briers et al., 2011). Weak organic acids, such as citric acid and malic acid, show a similar chelating activity, and malic acid shows an even more marked effect on outer membrane than citric acid(Oliveira et al., 2014).
In a previous study from our laboratory, a phage known as Bp7, with a wide host range, was isolated from chicken feces(Zhang et al., 2013). It encodes an endolysin, gp18(e)(further abbreviated as Bp7e), which has 76% homology with T4 lysozyme(data not shown). As a model microorganism, phage T4 has been well studied. In the study by Baase et al.(2010), T4 endolysin mutants were produced in order to study the relations between the structures and stabilities of these proteins; the results showed that the endolysin protein is very tolerant of change. The stability of T4 endolysin was associated with "large to small" substitutions of hydrophobic residues in the core, larger interior hydrophobic side-chains replaced with smaller ones lead a larger cavity volume and a loss stability of endolysin. The larger the cavity volume, the greater the loss of protein stability(Xu et al., 1998). T4 endolysin mutant L99A has the potential to cause a larger cavity volume, but with a lower protein stability(Liu et al., 2008). T4 endolysin mutant M102E may improve protein stability(Eriksson et al., 1992). However, the antibacterial activities of mutant L99A or mutant M102E have not been reported. In this work, protein Bp7e and its mutant Bp7Δe were expressed and their activities against Gram-positive bacteria were recorded; in addition, their activities against E. coli, S. paratyphi, or P. aeruginosa in the presence of permeabilizer were studied. We intended: i)to characterize a new T4-like coliphage lysozyme(Bp7e) and ii)to develop a much more efficient antibacterial protein by introducing mutations L99A and M102E.
-
Phage Bp7 was isolated by our research team and identified as a coliphage. Its complete genomic sequence was deposited in GenBank(HQ829472). Two Gram-positive strains and sixteen Gram-negative stains used in this report were kept in our lab; these strains are described in more details in Table 1. M. lysodeikticus was cultured in Luria-Bertani medium(LB, Takara, Dalian, China)at 30 ℃, and other bacterial strains were cultured in the same medium at 37 ℃. All bacterial strains were stored in 30% glycerol at -80 ℃ before use. Hen egg white lysozyme(HEWL, CW Biotech, China)was purchased from the specified suppliers.

Table 1. Host range of phage Bp7 and lytic activity of Bp7e and its mutation Bp7Δe
-
The Bp7e amino acid sequence was compared with that described in the NCBI GenBank database(http://www.ncbi.nlm.nih.gov/BLAST), and the phylogenetic relationships of Bp7e were analyzed using Mega5.0 software(http://www.megasoftware.net/index.php). The Bp7e secondary structure was analyzed using PredictProtein(http://www.predictprotein.org).
-
The Bp7e gene was amplified by PCR from a phage Bp7 genomic DNA template by a formard primer Bp7eF and a reverse primer Bp7eR containing Nde I and Xho I restriction sites, respectively(underlined in Table 2). The primers used in this work are listed in Table 2. The PCR product was purified and digested by Nde I and Xho I enzymes(Takara, Dalian, China) and cloned in a pET28a expression vector(Novagen, Shanghai, China)with a C-terminal 6×His tag. The presence of the insert in the recombinant plasmid pET28a-Bp7e was confirmed by DNA sequencing(Takara, Dalian, China).

Table 2. Sequences of PCR primers used for bacteriophage gene cloning.
The construct pET28a-Bp7e plasmid was transformed into E. coli BL21(DE3) and cultured on an LB plate with 50 μg/mL of Kana+ selection, and further confirmed by enzyme digestion, PCR and sequencing. The E. coli BL21(DE3)harboring the pET28a-Bp7e plasmid was cultured in 3 mL LB medium at 37 ℃ with 50 μg/mL of Kana+ to an optimal density of 0.6(OD600). Recombinant protein expression was induced for 12 h at 16 ℃ by the addition of isopropyl β-D-1-thiogalactopyr-anoside(IPTG)to a final concentration of 0.7 mmol/L. The culture was then centrifuged(4000 rpm, 10 min) and the cells were treated ultrasonically for 30 min(3 s pulse, 1 s pause)whilst being kept on ice. Samples of the supernatant and the precipitate were collected, and the presence of the recombinant proteins was confirmed by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis(SDS-PAGE). Insoluble cell debris was removed by centrifugation(10, 000 rpm, 3 min, 4 ℃). The recombinant protein in the supernatant was purified using a 6× His-tagged Protein Purification Kit(CW Biotech)as described by the manufacturer, and detected by western-blot analysis with an anti-His tag monoclonal antibody as the primary Ab and an HRP-conjugated rabbit anti-mouse IgG as the secondary Ab. The protein concentration was determined using a BCA Protein Assay Kit with bovine serum albumin(BSA)as st and ard(CW Biotech).
-
Two amino acid sites(L99A and M102E)of Bp7e were mutated using a Muta-directTM kit, following the prescribed protocol(SBS Genetech Co., Ltd.). Two sets of overlapping mutagenic primers – Bp7Δe 99F and Bp7Δe 99R for L99A, and Bp7Δe 102F and Bp7Δe 102R for M102E – are presented in Table 2(mutation basepairs are shown as shaded bases). Plasmid pET28a-Bp7Δe was then introduced into competent E. coli BL21(DE3)for heterologous production, as described above.
-
The antibacterial activity of Bp7e and Bp7Δe was assayed by measuring change in turbidity. Briefly, recombinant E. coli BL21(DE3)with plasmid pET28a-Bp7e(Group Bp7e)or pET28a-Bp7Δe(Group Bp7Δe)was grown to exponential stage in LB at 37 ℃ and then 0.7 mmol/L isopropyl-β-D-thiogalactopyranoside(IPTG)was added to the medium to induce expression. E. coli BL21(DE3)with plasmid pET28a was used as a negative control. The increase in the OD600 value of both test and control cultures was recorded every 1 h for 5 h. After 5 h induction, the cultures were then adjusted to the same OD600value, and a 100 μL sample of each culture was transferred to a 96-well plate. Then, 0.5 μL chloroform, which could work on outer membrane of E. coli and help the lysis effect of endolysin appear, was added per well and the fall in the OD600value of each well was recorded every 30 min for 3 h(Legotsky et al., 2014). All assays were performed in triplicate. The intracellular lysis activities of endolysin Bp7e and its mutant Bp7Δe were evaluated according to the changes in OD600.
-
The in vitro antibacterial activities of Bp7e and Bp7Δe were evaluated using different strains, as listed in Table 1, which includes two Gram-positive strains(S. aureus and M. lysodeikticus) and sixteen Gram-negative strains(fourteen E. coli strains, one S. paratyphi strain and one P. aeruginosa strain). In brief, the Gram-positive bacterial strains, cultured in LB to exponential stage, were harvested by centrifugation and resuspended in 10 mmol/L Hepes-HCl(pH 7.2)to a final concentration of 106 CFU/mL. To detect the antibacterial activities, 100 μL of Bp7e or Bp7Δe protein, at 1 μmol/L or 2 μmol/L, respectively, was mixed with 100 μL of bacterial suspension and incubated at room temperature for 2 h. After incubation, a serial, 10-fold dilution series in phosohate-buffered saline(PBS)was performed for each sample, and 100 μL of each dilution was plated on LB agar plates. After overnight incubation, the number of viable bacteria was assessed by counting the colonies that had formed in the LB agar. PBS(pH 7.2)was used as a negative control instead of Bp7e and Bp7Δe protein, and HEWL(2 μmol/L)was used as a positive control. All assays were performed in triplicate.
The antibacterial activities of the Bp7e and Bp7Δe proteins against Gram-negative bacterial strains in the presence of malic acid were also assessed. Briefly, the bacterial strains cultured at the exponential phase were harvested by centrifugation and resuspended in 10mmol/L Herpes-HCl(pH 7.2)to a final concentration of 106 CFU/ mL. To test the antibacterial activity of Bp7e and Bp7Δe, samples with the following composition were prepared:(ⅰ)100 μL of bacterial suspension, 50 μL of 20 mmol/L malic acid and 50 μL of Bp7e or Bp7Δe protein(2 μmol/L; 4 μmol/L); (ⅱ)100 μL of bacterial suspension, 50 μL of PBS and 50 μL of Bp7e or Bp7Δe protein(2 μmol/L; 4 μmol/L); (ⅲ)100 μL of bacterial suspension, 50 μL of PBS and 50 μL of HEWL(2 μmol/L; 4 μmol/L); (ⅳ)100 μL of bacterial suspension, 50 μL of 20 mmol/L malic acid and 50 μL of HEWL(2 μmol/L; 4 μmol/L); (ⅴ)100 μL of bacterial suspension, 50 μL of 20 mmol/L malic acid and 50 μL of PBS(pH 7.2);(ⅵ)100 μL of bacterial suspension and 100 μL of PBS(pH 7.2). All samples were incubated at room temperature for 30 min. After incubation, a serial, 10-fold dilution series in PBS was performed for each sample, and 100 μL of each dilution was plated on LB agar plates. After overnight incubation at 37 ℃, the number of viable bacteria was assessed by counting the colonies that had formed in the LB agar. Here, PBS, rather than malic acid or protein(Bp7e, Bp7Δe and HEWL), served as a negative control; HEWL(2 μmol/L or 4 μmol/L)served as a positive control. All assays were performed in triplicate.
The antibacterial activity against the eighteen bacterial strains was quantified as the relative inactivation in logarithmic units = log10(N0/N1), where N0 is the colony number of the untreated group(in the negative control) and N1 is the colony number of the treated groups after incubation. Averages ± st and ard deviations were calculated for n = 3 repeats. All the data were analyzed by SPSS 11.0 statistical software(SPSS, Chicago, IL, USA).
Bacteria, phage and chemicals
Sequence analysis of Bp7e
Cloning, protein expression and purification of Bp7e
Site-directed mutagenesis of Bp7e
Intracellular lysis activity assay of Bp7e and Bp7Δe
Antibacterial activity assay of Bp7e and Bp7Δe
-
The 489 bp endolysin gene(Bp7e)of coliphage Bp7, encoding a 162-amino acid protein with a deduced molecular mass of 19.6 kDa, was amplified and sequenced. Next, a bioinformatic analysis was conducted to analyze the primary and secondary structures of Bp7e. Primary structure comparisons of Bp7e using BlastP software revealed a very high level of homology( > 98%)with endolysins JS98 and IME08, and with eight other endolysins from Enterobacteriaceae phages – namely, vB EcoM VR7, SP18, vB EcoM JS09, RB69, vB EcoM PhAPEC2, T4, RB14 and RB32. All of the homologies were greater than 76%.The phylogenetic relationships among 11 endolysins of the phages mentioned above are shown in Figure 1. Bp7e was predicted to be a global protein with a conserved domain between amino acids 3 and 161 and belonged to a lysozyme-like superfamily. This class of enzymes degrades the cell wall by hydrolyzing the β-1, 4 linkages between N-acetylmuramic acid and N-acetyl-D-glucosamine residues(Fischetti, 2008). Secondary structure analysis using PredictProtein revealed no disulfide bond in the Bp7e protein, even where there were two cysteine residues. Our study found eight α-helices(51.85%), three β-sheets(8.02%) and loops(40.12%).
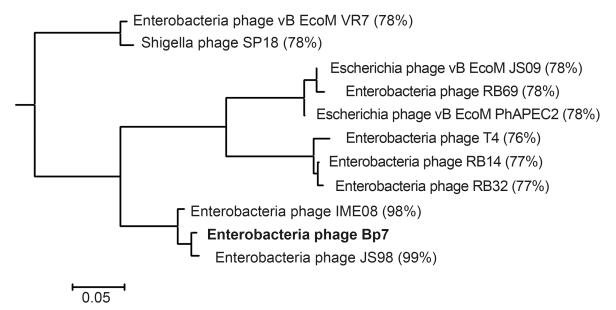
Figure 1. Homology analysis of Bp7 endolysin.The phylogenetic relationships among 11 endolysins of Enterobacteriaceae phages were analyzed.Bp7e showed more than 76% homology to other endolysins that belonged to a T4-like genus, and were closely related to JS98 and IME108( > 98%).
-
The 489 bp of the Bp7e gene was amplified and inserted into expression vector pET28a to construct the pET28a-Bp7e recombinant plasmid. The construct was characterized by digesting with Nde I and Xho I, and AGE(agarose gel electrophoresis)results indicated that pET28a-Bp7e was correctly constructed, which was further verified by sequencing analysis(data not shown).
Using the pET28a-Bp7e plasmid as a template, two amino acid sites were mutated by two PCR reactions(L99A and M102E), and the pET28a-Bp7Δe mutant was produced. This was also verified by sequencing analysis(data not shown).
-
The E. coli BL21(DE3)harboring either pET28a-Bp7e or pET28a-Bp7Δe was cultured in kana+ LB and induced as described previously. The expressed products were analyzed by SDS-PAGE and western blot analysis(Figure 2). The results showed that His-tagged Bp7e and Bp7Δe were mainly expressed in the supernatant and represented the desired protein of 22 kDa. Overexpression of Bp7e and Bp7Δe in E. coli BL21(DE3)yielded soluble proteins with > 41.6% and 72.1% purity, respectively.
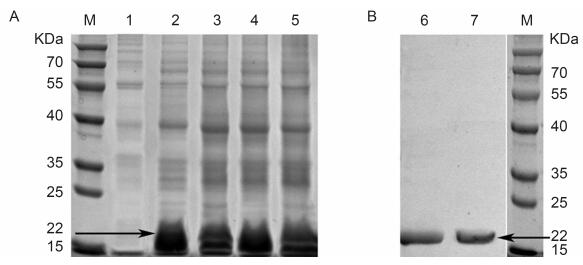
Figure 2. Protein expression and purification of Bp7e and Bp7Δe.(A) E.coli BL21(DE3) cultures harboring the backbone plasmid of pET28a or recombinant plasmid (pET28a-Bp7e, pET28a-Bp7Δe) were induced with 0.7 mmol/L IPTG. After all the cells were lysed, the protein samples in the supernatant and precipitation were collected and assessed by SDS-PAGE.(B) The recombinant endolysin Bp7e and Bp7Δe (~22 kDa) were purified by Ni-affinity chromatography and detected by western blot analysis with an anti-His tag monoclonal antibody as the primary Ab and HRP-conjugated rabbit anti-mouse IgG as the secondary Ab.M:protein molecular weight markers; 1:control; 2:Bp7e protein in supernatant; 3:Bp7e protein in precipitate; 4:Bp7Δe protein in supernatant; 5:Bp7Δe protein in precipitate; 6:western blot analysis of Bp7e;7:western blot analysis of Bp7Δe.
-
Bacteriophage Bp7 was able to infect and lyse the E. coli BL21(DE3)lab strain(data not shown), so the intracellular lysis activities of the Bp7e and Bp7Δe proteins were assayed by measuring the changes in turbidity ay. BL21(DE3)with plasmid pET28a(control), pET28a-Bp7e, and pET28a-Bp7Δe were cultured in kana+ LB. After IPTG was added to induce protein expression, the OD600 values of BL21(DE3)with Bp7e or Bp7Δe plasmids were found to increase at a slower rate than those of the control. After 5 hours of IPTG induction, the OD600 values of the control reached about 0.8; however, the OD600 values of the cells expressing Bp7e or Bp7Δe were less than 0.6. No obvious difference was found between the Bp7e and Bp7Δe groups within 5 h of induction(Figure 3A). Next, the culture turbidity of all of the groups was adjusted to the same level and treated with 0.5%(v/v)chloroform, and the lysis activity of the Bp7e and Bp7Δe proteins was evaluated. The results showed that the OD600 value of the control remained stable(at about 1.0)over the subsequent 2 hours, while the values of the Bp7e and Bp7Δe groups showed an obvious reduction(at about 0.85)(Figure 3B).
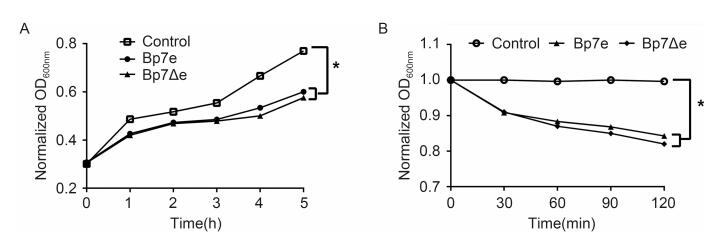
Figure 3. Intracellular lysis activity of Bp7e and Bp7Δe.0.7mmol/L IPTG was used to induce the expression of Bp7e and Bp7Δe in E.coli BL21(DE3).The culture turbidity of all the groups was assayed every 1 h for 5 h.The OD600 values of the Bp7e and Bp7Δe groups increased at a slower rate than the control group (A).The culture turbidity of all the groups was adjusted to the same level after IPTG induction for 5 h, and the intracellular lysis activities of the Bp7e and Bp7Δe proteins were assayed by measuring the changes in turbidity in the presence of 0.5% chloroform.The OD600 value of the control group remained stable over the subsequent 2 hours, but the OD600 values of the Bp7e and Bp7Δe groups showed an obvious decrease (B).*showed a significant OD600 value difference compared with the control group at 5 h (P < 0.05).
-
The in vitro antibacterial activities of Bp7e and Bp7Δe proteins against S. aureus and M. lysodeikticus were assayed(Figure 4). M. lysodeikticus, as a Gram-positive bacterium, was sensitive to both Bp7e and Bp7Δe, as expected(P < 0.05). The cell number showed an obvious decrease after treatment by Bp7e or Bp7Δe at 1 μmol/L for 2 h, and the antibacterial activities of Bp7e or Bp7Δe at 2 μmol/L were much higher than those at 1 μmol/L after 2 h of incubation(P < 0.05). Bp7Δe showed a higher antibacterial activity against M. lysodeikticus than Bp7e(P < 0.05)– 2 μmol/L Bp7Δe could cause about 0.7 log reduction in viable cell number – however, natural endolysin Bp7e at 2 μmol/L resulted in only an approximate 0.4 log reduction in viable cell number. Compared with HEWL, Bp7e and Bp7Δe showed much lower activities(P < 0.05). Bp7e and Bp7Δe showed a negative antibacterial activity against S. aureus. Overall, Bp7Δe protein demonstrated a higher antibacterial activity against M. lysodeikticus than Bp7e, but a much lower activity than HEWL.
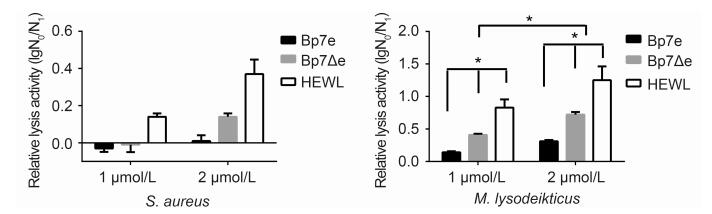
Figure 4. Antibacterial activity of Bp7e and Bp7Δe against S.aureus and M.lysodeikticus.The antibacterial activity of Bp7e or Bp7Δe against S.aureus (A) or M.lysodeikticus (B) at 1 μmol/L and 2 μmol/L concentrations, respectively, were examined at 2 h.PBS was used as a negative control instead of endolysin proteins.HEWL at the same concentration was used as a positive control.The antibacterial activity was shown as the relative inactivation in logarithmic units (=log10(N0/N1), where N0 is the number of untreated cells and N1 is the number of cells after treatment).Averages ± standard deviations were calculated for n=3 repeats.*showed a significant log reduction unit compared with the PBS group or an obvious difference between groups (P < 0.05).
The antibacterial activities of Bp7e and Bp7Δe against sixteen Gram-negative bacterial strains were also assayed in the presence of malic acid(Figure 5). These Gram-negative bacterial strains included fourteen E. coli strains(nine of which could be lysed by phage Bp7), one P. aeruginosa strain and one S. paratyphi strain. As expected, in the absence of malic acid no antibacterial activities of endolysin Bp7e, Bp7Δe and HEWL against E. coli cells were detected because the outer membrane of E. coli prevented the proteins from reaching the peptidoglycan layers. In the presence of malic acid, Bp7e and Bp7Δe showed an obvious antibacterial activity against certain bacterial strains, and their activities continued to rise with increasing protein concentration. Bp7e and Bp7Δe showed a negative activity against P. aeruginosa or S. paratyphi(P > 0.05); among the nine host E. coli strains of phage Bp7, seven E. coli strains were sensitive to both Bp7e and Bp7Δe. Moreover, Bp7Δe at 4 μM could lyse one additional host strain(EZ041122A)of phage Bp7(P < 0.05), while Bp7e had an indistinct activity on this strain, indicating that the site mutations(L99A, M102E)could improve the activity of endolysin Bp7e. In addition, all of the proteins showed a higher lysis activity at 4 μmol/L than at 2 μmol/L, indicating that higher concentrations lead to stronger antibacterial activities. The E. coli strain(C041027A), as the host of phage Bp7, showed resistance to Bp7e and Bp7Δe, but in the presence of malic acid 4 μmol/L Bp7Δe treatment resulted in an obvious reduction in cell number compared with 2 μmol/L Bp7Δe treatment(P < 0.05). The other five E. coli strains that were not the host of phage Bp7 were also resistant to Bp7e and Bp7Δe. All the results showed that the lysis range of endolysin and its mutant was similar to that of parental phage Bp7. Bp7e did not show a broader lysis range than phage Bp7, even when site mutations were introduced into endolysin Bp7e. Compared with Bp7e and Bp7Δe, HEWL was able to lyse more Gram-negative bacterial strains, such as E. coli CH041025A and P. aeruginosa. However, it could not lyse all the Gram-negative strains: five E. coli strains and one S. paratyphi strain showed resistance to HEWL.
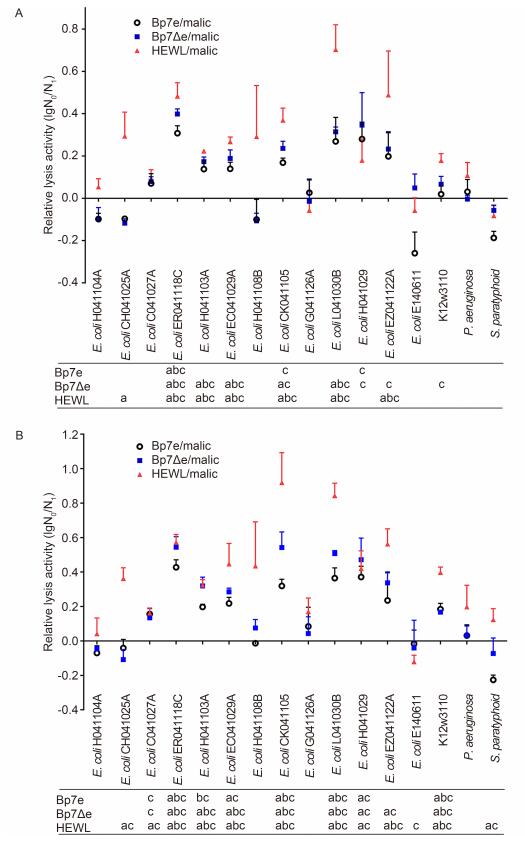
Figure 5. Antibacterial activity of Bp7e and Bp7Δe against Gram-negative bacterial strains in the presence of malic acid.The antibacterial activities of Bp7e and Bp7Δe against E.coli at 2 μmol/L (A) and 4 μmol/L (B) concentrations were examined in the presence of malic acid (5 mmol/L).Bacterial incubation with protein/PBS or malic acid/PBS served as a negative control, while HEWL at the same concentration was used as a positive control.The antibacterial activity of the protein was quantified as the relative inactivation in logarithmic units (=log10(N0/N1), where N0 is the number of untreated cells and N 1 is the number of cells after treatment).Averages ± standard deviations were calculated for n=3 repeats.After incubation, significant log reduction units compared with the malic acid group were indicated in a (P < 0.05), significant log reduction units compared with the PBS group were indicated in b (P < 0.05), and significant log reduction units compared with the endolysin without malic acid group were indicated in c (P < 0.05).
In all of the tested E. coli strains, seven of the fourteen strains were all sensitive to Bp7e, Bp7Δe and HEWL, and no significant difference was found in the antibacterial activities of these proteins(P > 0.05). Moreover, E. coli strain EZ041122A was much more sensitive to HEWL and Bp7Δe than Bp7e(P < 0.05), and HEWL showed a much higher level of lysis activity against E. coli strain C041027A than Bp7Δe and Bp7e(P < 0.05). These results indicate that there are differences in the lysis activities of endolysin Bp7e or Bp7Δe against different host bacterial strains.
In brief, Bp7e and Bp7Δe were able to lyse most host strains of phage Bp7 in the presence of malic acid, and also possessed antibacterial activity against M. lysodeikticus. Bp7Δe protein with double-site mutations exhibited a stronger antibacterial activity and a broader host range of E. coli strains than Bp7e.
Sequencing and analysis of Bp7e
Construction and identification of pET28a-Bp7e and pET28a-Bp7Δe
Expression and purification of proteins Bp7e and Bp7Δe
Intracellular lysis activity assay of Bp7e and Bp7Δe
Antibacterial activity assay of Bp7e and Bp7Δe
-
Chicken colibacillosis is a worldwide bacterial disease which brings huge economic loss in China, and E. coli is the major pathogen of this disease(Olsen et al., 2012). Antibiotics have been widely used to control this disease, but these cause serious multi-resistance problems(Dupont et al., 2007; Zhang et al., 2012; Xu et al., 2015). Phage-encoded endolysins have recently been considered as a potential agent for treating clinical bacterial infections, which are believed to have a broader lytic spectrum than phages(Borysowski et al., 2006). Moreover, the development of resistance against endolysins has not been reported(Burrowes et al., 2011; Li et al., 2012). In this research, we analyzed the antibacterial activity of Bp7e, an endolysin produced by phage Bp7, to explore its potential application in controlling bacterial diseases, especially the virulent E. coli infection. Furthermore, the endolysin mutant Bp7Δe was prepared, in order to study the change in antibacterial activity following mutation.
Numerous endolysin genes of bacteriophages have been determined in previous reports. Phage Bp7e has been shown to share more than 76% homology with ten other predicted or sequenced phage endolysins, especially with endolysins JS98 and IME08(98%). Among the ten endolysins, most were produced by a T4-like phage group of Enterobacteriaceae phages. Bp7e was found to share a high degree of homology with these endolysins, a finding that was consistent with that of an earlier study which found that phage Bp7 belongs to a T4-like phage group(Zhang et al., 2013). It has also been reported that endolysin of T4 and HEWL can break the β-(1, 4)-glycosidic bonds in the peptidoglycan of the Gram-positive bacteria cell wall and cause bacteriolysis, but it had little effect on Gram-negative bacteria because the peptidoglycan was sequestered beneath the outer membrane(Lukacik et al., 2013). Many studies have investigated the destruction of Gram-negative bacteria. Oliveria et al.(2014) reported an endolysin(Lys68)produced by Salmonella phage and its potential antimicrobial effectiveness when combined with organic acids against Gram-negative pathogens. The paper demonstrated that Lys68 was able to lyse a wide spectrum of Gram-negative bacteria in combination with the outer-membrane permeabilizers EDTA, citric acid and malic acid. In our previous study, EDTA and citric and malic acids were all used as permeabilizers to assay the antibacterial activity of Bp7e and Bp7Δe proteins against E. coli Malic acid was demonstrated to be the most effective permeabilizer(data not shown), so it was used to study the antibacterial effectiveness of Bp7e and Bp7Δe in this work. Bp7e was shown to inhibit the growth of Gram-positive M. lysodeikticus, and exhibited antibacterial activity against E. coli in combination with malic acid. In addition, another study reported that endolysin has a broader lytic spectrum than phages(Borysowski et al., 2006), but in this work the lytic spectrum of Bp7e was narrower than the host range of phage Bp7.
According to previous studies, the lysozyme structure shows a cavity in the core of the T4 lysozyme which can combine with lig and s on the host cell surface and initiate the infection(Baase et al., 2010). The cavity size is associated with lysozyme stability. Point mutations cause very modest, localized changes in the structure(López et al., 2013). Moreover, it has been reported that the L99A mutation, which is located in the cavity of the T4 lysozyme, can result in a largely empty cavity that benefits lig and combination but causes a reduction in stability(Eriksson et al., 1992; Liu et al., 2008). Recent studies have shown that this cavity can retain its shape and volume under high pressure. Another mutation M102E, which is located in the wall of the cavity of the T4 lysozyme, can cause the lysozyme to bind both polar and nonpolar lig and s, because this mutation results in the binding of three water molecules close to Glu102(Liu et al., 2009). These reports described the influence of site mutation on the T4 lysozyme structure, but the effects of these mutations on its antibacterial activities have not yet been reported. In this work, we focused on the relationship between site mutations and endolysin lysis activities. The endolysin Bp7e, produced by phage Bp7, was double-site mutated with L99A and M102E, and it was demonstrated that the L99A/M102E mutation had a broader lytic spectrum and exhibited better antibacterial activities than the Bp7e wild type.
In conclusion, the phage endolysin Bp7e was shown to exhibit antibacterial activity against M. lysodeikticus and seven E. coli strains in the presence of malic acid, and the L99A/M102E mutation of Bp7e was shown to lyse one additional E. coli strain and thus showed improved antibacterial activity.
-
We would like to thank Prof. Qigen Tong, Dept. of Food Science, Beijing University of Agriculture, for his gift of M. lysodeikticus. Funds for this research were provided by the Natural Science Foundation of Shan-dong Province, China(ZR2015CM020 and ZR2013CQ024).
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
HR conceived the studies and participated in experimental design and coordination. CZ, YW and HS carried out the experiments. CZ analyzed the data and wrote the paper. All authors read and approved the final manuscript.

 DownLoad:
DownLoad: