












-
Dear Editor,
As a member of the Kobuvirus genus in the Picornaviridae family, porcine kobuvirus is prevalent worldwide in pigs with or without diarrhea. Picornaviruses are classified into 12 genera(Aphthovirus, Cardiovirus, Enterovirus, Hepatovirus, Kobuvirus, Parechovirus, Sapelo-virus, Avihepatovirus, Erbovirus, Senecavirus, Tescho-virus, and Tremovirus)(Reuter et al., 2011). Kobuvirus contains two officially recognized species, Aichi virus and bovine kobuvirus(Carmona-Vicente et al., 2013). Porcine kobuvirus was first identified in 2007 in stool specimens in Hungary(Reuter et al., 2008) and has also been found in several countries, such as Thailand (Yu et al., 2009), Brazil(Park et al., 2010), South Korea(An et al., 2011), the Netherlands(Barry et al., 2011), Japan(Okitsu et al., 2012), and the United States of America(USA)(Sisay et al., 2013). However, the virus has been reported in very few districts of China, such as Shanghai(Wang et al., 2011) and Gansu(Fan et al., 2013). In this study, we present the development of a reverse transcription polymerase chain reaction(RT-PCR)assay to detect porcine kobuvirus and describe the results of epidemiological study and variation analysis of the porcine kobuvirus 3D gene in Sichuan province, China.
In total, 200 stool specimens were collected as suspected disease materials from 23 farms during February 2012 to April 2015, including nine cities in Sichuan province(Mianyang, Zigong, Suining, Ganzi, Deyang, Chengdu, Guang'an, Ya'an, and Meishan). One hundred and fifty specimens were from pigs with diarrhea, and 50 specimens were from pigs without diarrhea. Of these, 95, 65, and 40 specimens were from suckling pigs(less than 4 weeks of age), weaned pigs(less than 7 weeks of age), and growing pigs(over 7 weeks of age), respectively. Stool specimens were converted to 10%(w/v)suspensions in phosphate-buffered saline(PBS), and total RNA was extracted from 200-μL suspensions using an RNA extraction kit. All samples were detected by RT-PCR with specific primers(P1: 5'-CGTCTCATAGGAGA CGAACG-3' and P2: 5'-TTTGTCGTAGAACTCCTT GA-3'), which were designed to target regions of the porcine kobuvirus 3D gene in the S-1-HUN and K-30-HUN strains.
Our analysis showed that 60%(120/200)of samples were positive for porcine kobuvirus, indicating that the virus was ubiquitously distributed throughout Sichuan province. However, the prevalence rates in different districts varied dramatically, reaching 85.71%(18/21)in Deyang, 83.33%(20/24)in Suining, 73.68%(14/19)in Meishan, 65.38%(17/26)in Chengdu, 59.25%(16/27)in Zigong, 56.52%(13/23)in Mianyang, 46.67%(14/30)in Ya'an, 31.25%(5/16)in Ganzi and 21.43%(5/16)in Guang'an. The prevalence rates decreased with age: 59.17%(71/120)of cases observed in suckling pigs, 27.50%(33/120)of cases in weaned pigs, and 13.33%(16/120)in finishing pigs(Supplementary Table S1). Statistical analysis showed that suckling pigs were far more susceptible to porcine kobuvirus infection than weaned pigs and finishing pigs. Therefore, our analysis showed that porcine kobuvirus infection was a major cause of diarrhea in piglets.
Next, we performed variation analysis of the porcine kobuvirus 3D gene in 23 representative samples selected from 120 positive samples. After sequencing, the new sequences were submitted to GenBank with accession numbers JX164113-JX164135. We analyzed the homology of these sequences and compared the homologies of these strains with 26 reference strains from GenBank. The nucleotide and amino acid identities among the 23 new sequences were 92.6%-99.6% and 96.8%-98.9%, respectively. The nucleotide identities between the new and reference sequences were 91.1%-96.8%, and the amino acid identities were 92.6%-99.6%. Therefore, there were no obvious differences in the amplified sequences, suggesting that the porcine kobuvirus 3D sequence was relatively conserved.
All 49 sequences were used to construct a phylogenic tree by the neighbor-joining(NJ)method with MEGA 6.0(Figure 1A). The results showed that the global porcine kobuvirus strains could be divided into two distinct groups(A and B) and that all new Sichuan strains belonged to group A. The majority of porcine kobuvirus strains from China and other countries were classified into group B. Specifically, 14 new Sichuan strains formed a close cluster in subgroup A1, with nucleotide identities of 93.7%-99.6%. Additionally, six new strains belonged to subgroup A2, with nucleotide identities of 97.9%-99.3%. The WUH1, SC, and BRA74 strains, in addition to three new strains, were distributed into subgroup A3, with nucleotide identities of 91.5%-97.9%. In subgroup B1, the homology of the SH-W-CHN, JS-02a-CHN, SwKoV CH441, and JS-01-CHN isolates was 93.7%-96.5%, and all isolates were identified recently in China. The subgroup B2 included the CMP06/07-THA isolate in Thailand, the OH/RV50 isolate in the USA, the HUN isolates in Hungary, three South Korean isolates, and 11 Chinese isolates, with nucleotide homology of 90.4%-96.4%. The homologies of the 23 new strains identified in this study were obviously more similar to strains isolated previously in China than to those in Hungary, Brazil, South Korea, Thailand, and other countries.
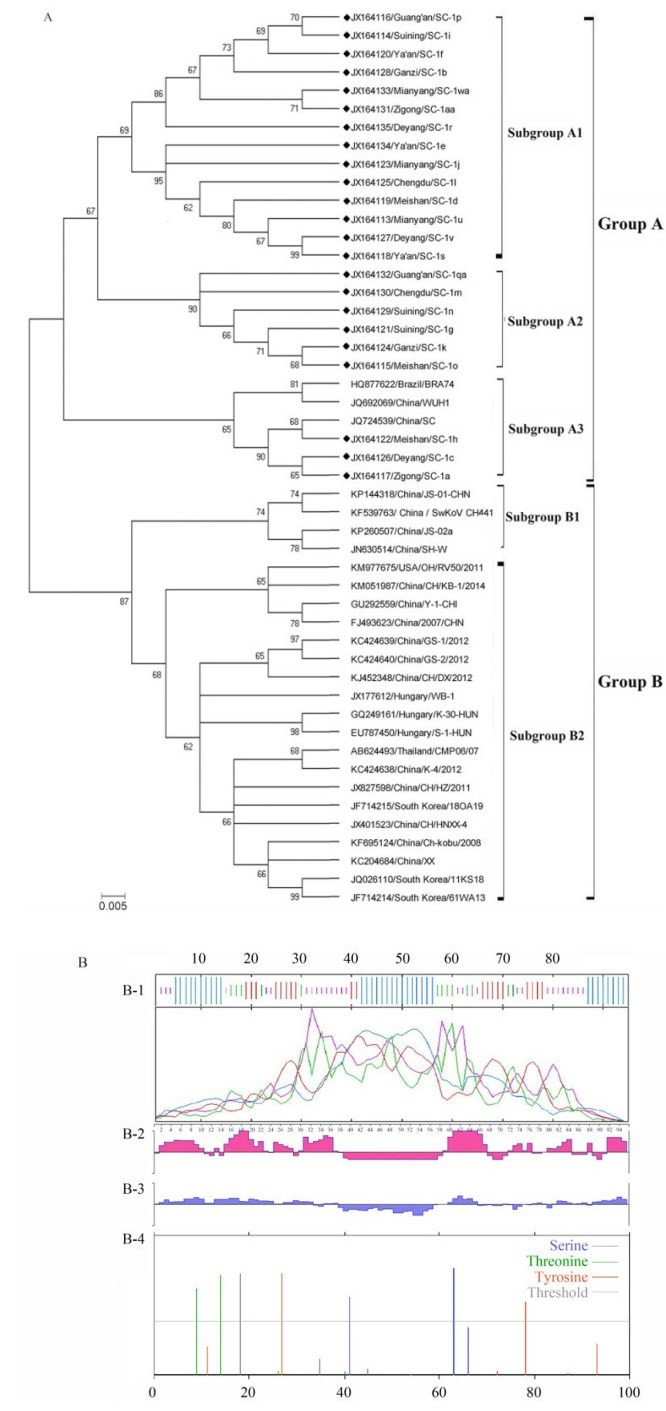
Figure 1. Variation analysis of porcine kobuvirus 3D gene. (A) Phylogenetic analysis of porcine kobuvirus 3D patial sequences between new Sichuan strains and reference strains from GenBank. The phylogenetic tree was constructed by the neighbor-joining method and Kimura 2-parameter model. The sequence particulars were shown as Accession number/region/Strain. Filled quadrate (◆) indicates porcine kobuvirus 3D protein sequences collected in this study. (B) Illustration of the porcine kobuvirus 3D patial protein detected in this study. B-1: Analysis of the secondary structure. The a-helix, β-turn, the extended strand and the random coil are denoted by blue, green, red and purple curves, respectively; B-2: Prediction of antigenic epitope; B-3: Hydrophilicity Plot; B-4: Analysis of phosphorylation site.
Nucleotide and amino acid analyses of 23 Sichuan strains were also performed in this study. New nucleotide sequences were found at 40 mutational sites compared with the 61WA13 strain in South Korea and the S-1-HUN strain in Hungary, including nucleotides at positions 6, 9, 15, 33, 39, 48, 51, 54, 63, 69, 72, 93, 96, 102, 114, 117, 120, 126, 129, 132, 141, 144, 147, 148, 159, 168, 171, 186, 189, 192, 198, 201, 207, 208, 213, 231, 243, 252, 267, and 273, of which 28 mutations were found in more than one sequence. For prediction of the secondary structure by SOPMA, several sites were concentrated at the alpha helix(35.79%), beta turn(13.68%), random coil(29.47%), and extended strand (21.05%)in the amino acid sequence(Figure 1B-1). The T-cell epitope prediction with Protean in DNAstar was mainly concentrated in the regions of amino acid positions 2-37, 59-67, and 91-95(Figure 1B-2), roughly consistent with the prediction of hydrophilic sites(Figure 1B-3). Additionally, the 49 global strains had seven common amino acid substitutions(I3→T, E6→V, T9→A/V, N24→K, C37→R, I43→V, and I50→V), most of which were concentrated in the 3D epitope regions. Analysis of the phosphorylation sites by NetPhos 2.0 showed that there were three serine phosphorylation sites(S18, S41, and S63), two threonine phosphorylation sites(T9 and T14), and two tyrosine phosphorylation sites(Y27 and Y78), with a threshold value of 0.5(Figure 1B-4). The maximum likelihood of transition/ transversion propensity and substitution matrix of the amplified nucleotides were also estimated by MEGA 6.0(Table 1).
Chinese porcine kobuvirus strains Global porcine kobuvirus strains A T/U C G A T/U C G A - 1.94 1.78 6.07 A - 1.97 1.80 7.25 T/U 1.70 - 34.89 1.41 T/U 1.71 - 33.54 1.43 C 1.70 38.10 - 1.41 C 1.71 36.71 - 1.43 G 7.29 1.94 1.78 - G 8.67 1.97 1.80 - Analysis of the new Sichuan porcine kobuvirus strains, reference Chinese strains and global strains. Each entry is the probability of substitution (r) from one base (row) to another base (column). The rates of different transition substitutions are shown in bold and those of transversion substitutions are shown in italics. Table 1. Maximum likelihood estimate of substitution matrix
Our study indicated that porcine kobuvirus was widely prevalent in Sichuan province, China. The prevalence in diarrhea specimens(75.83%, 91/120)was similar to that observed in Brazil(78.4%)(Barry et al., 2011), but lower than those in Korea(84.5%)(Park et al., 2010) and Thailand (97%)(Khamrin et al., 2009). In pigs without diarrhea, the infection rate(24.17%, 29/120)was similar to that in Shanghai(22.4%)(Wang et al., 2011) and the USA(21.7%)(Verma et al., 2013), but obviously lower than that in Hungary(65%)(Reuter et al., 2010). In addition, Chen reported that the prevalence rates of porcine kobuvirus in southwest China were 64.3% and 29.4% in pigs with and without diarrhea, respectively(Chen et al., 2013), which was slightly inconsistent with our current research. Additional studies are required to confirm whether porcine kobuvirus contributes to the pathogenesis of diarrhea and to determine its specific pathogenic mechanisms in piglets.
As a viral RNA-dependent polymerase, porcine kobuvirus 3D protein is involved in regulating viral RNA replication and plays an important role in virus proliferation(Verma et al., 2013). Therefore, we first conducted molecular and phylogenic analysis of the porcine kobuvirus 3D region. According to the phylogenic tree, the porcine kobuvirus 3D gene was relatively conserved, and the virus has not evolved dramatically since it was first isolated in 2007. A report by Chen showed that the nucleotide and amino acid identities of the porcine kobuvirus VP1 region range from 97.5%-98.8% and 98.2%-98.7%, respectively(Chen et al., 2013); this variability was less than that observed in our current study. Moreover, some point mutations were observed in the analyzed sequences; these mutations may have changed the conformation and function of the 3D protein. These current findings provide insights into the epidemiology and genetic characteristics of this newly emerging virus.
HTML
-
This work was supported by New Century Excellent Talents at the University of Ministry of Education of China(Project No: NCET-11-1059) and the International Science and Technology Cooperation Projects of the Ministry of Science and Technology(Project No. 2014DFA31260). All the specimens were reviewed and approved by the Institute of Animal Health Animal Care and Use Committee at Sichuan Agricultural University(approval number: SYXK2014-187). The authors declare that they have no competing interests associated with the work presented herein.
Supplementary Table S1 are available on the website of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-
Pig age diarrhea non-diarrhea Total Infection rates (%) Suckling pigs 56 15 71 59.17 Weaned pigs 24 9 33 27.50 Growing pigs 11 5 16 13.33 Total 91 29 120 - Infection rates (%) 75.83 24.17 - - Table S1. Statistical analysis of samples infected with porcine kobuvirus.

 DownLoad:
DownLoad: