












HTML
-
Nosocomial infections are a worldwide public health problem(Rosenthal et al., 2014). Staphylococcus aureus, a Gram-positive bacterium found in humans and animals, causes the most frequent infections(Méric et al., 2015). The symptoms of S. aureus infections range from relatively mild to life threatening(Łobocka et al., 2012). The intractable infections caused by methicillin-resistant S. aureus(MRSA)are particularly dangerous(Moellering, 2012). The identification and study of MRSA-virulent phages may have a significant medical impact in tackling the emergent MRSA threat(Kaur et al., 2012).
Phage life cycles depend largely on the processes involved in whole genome packaging, starting from initiation(Fujisawa and Morita, 1997)to viral DNA replication(Zaballos et al., 1988), termination and regulation of transcription(Stewart et al., 2009). These processes rely on identification of the genome termini for successful whole genome packaging. However, the mechanism of DNA end formation in MRSA-virulent Twortlikevirus phages is not clearly understood(Łobocka et al., 2012).
In this study, we isolated and analyzed four newly identified MRSA-virulent Twortlikevirus Staphylococcus phages, IME-SA1, IME-SA2, IME-SA118 and IME-SA119. By applying our proposed "termini analysis theory"(Zhang et al., 2015), we identified genome termini. BLAST results showed that these phages belong to the family Myoviridae, subfamily Spounavirinae, Twortlikevirus S. phage G1 species. Further analysis revealed that Twortlikevirus S. phage G1 and K species have conserved termini. Interestingly, a variable region close to the genome terminus was found in G1 species. This is the first report to identify the termini of Twortlikevirus S. phage using only High-throughput sequencing(HTS)data, and to discover the conserved terminus and its adjacent variable region.
-
We analyzed and identified the termini of the Twortlikevirus S. aureus phages IME-SA1, IME-SA2, IME-SA118 and IME-SA119 using our termini analysis theory, described previously(Zhang et al., 2015). In brief, this theory presumes that the HTS reads with the highest occurrence frequencies represent the termini of phages.
-
Four Twortlikevirus S. aureus phages, IME-SA1/2/ 118/119, were isolated from sewage in the 307th Hospital of Chinese People's Liberation Army(PLA hospital 307, Beijing, China), while the host bacteria for the four phages were isolated from clinical samples in the same hospital. The collaboration between PLA hospital 307 and our laboratory allowed us to carry out experiments with no special permit requirements. The samples collected were neither privately owned nor protected, and did not involve any endangered or protected species.
Enrichment cultures (Adams, 1959) were used to isolate IME-SA1/2/118/119 from sewage. Specifically, 4 mL of sewage sample was mixed with 2 mL of liquid LB medium(tryptone 30 g/L, yeast extract 15 g/L, and NaCl 30 g/L) and was incubated at 37 ℃ overnight. The culture was centrifuged at 13000 × g for 10 min and the supernatant was subsequently collected, mixed with host bacteria, and plated to obtain single bacteriophage plaques.
Approximately 5 mL of an overnight culture of host bacteria was mixed with 500 mL of fresh liquid LB medium and grown at 37 ℃ to log phase(OD = 0.6). At this point, IME-SA1/2/118/119 was added at a multiplicity of infection of 0.01(109 PFU bacteriophage per liter), and the culture was continued at 37 ℃ until the host bacteria were completely degraded. This occurred after 6 h. The concentration of IME-SA1/2/118/119 in culture was measured using standard protocols(Carlson, 2005). Then, DNase Ⅰ and RNase A were added to 500 mL of each culture to a final concentration of 0.1%, mixed gently and allowed to st and at room temperature for 30 min. Solid NaCl was added to this culture to a final concentration of 1 mol/L and incubated in an ice-water bath for 1 h. Then, the culture was centrifuged at 11, 000 × g for 10 min to remove cell debris, and polyethylene glycol 6000(PEG6000) was added to the supernatant to a final concentration of 10%(w/v)while slowly stirring at room temperature. This solution was transferred to a polypropylene centrifuge tube in an ice-water bath and incubated for at least 1 h to precipitate the phage particles. Following centrifugation(11, 000 × g for 10 min at 4 ℃), the phage-containing precipitate was resuspended in 8 mL of SM buffer. Each 1L SM buffer contains 5.8g NaCl, 2g MgSO4·7H2O, 50mL 1 mol/L Tris-Cl(pH7.5) and 5mL 2%(w/v)gelatin. The SM buffer incubated for 1 h at room temperature. An equal volume of chloroform was then added to separate the phage particles from the PEG6000. The mixture was centrifuged at 3, 000 × g for 10 min and the aqueous phase was recovered and filtered through a 0.22-μm pore-size membrane filter to remove debris. About 1 mL of each phage suspension was layered on cesium chloride gradient solutions(gradient 1 is 1.3 g/mL that is 0.45 g of cesium chloride in 1.0 mL of water; gradient 2 is 1.5 g/mL that is 0.83 g of cesium chloride in 1.0 mL of water; gradient 3 is 1.7 g/mL that is 1.28 g of cesium chloride in 1.0 mL of water)in 5.0 mL cellulose nitrate centrifuge tubes(Bachrach and Friedmann, 1971). After centrifugation in a Beckman Coulter Swinging Bucket Rotor(SW41, Ti)for 15 min at 3, 000 × g, the concentrated phages in a visible band were collected using a capillary pipette. The purified phage was stored at 4 ℃.
Genomic DNA from IME-SA1/2/118/119 was extracted based on a previously described method(Carlson, 2005). In brief, the purified phage IME-SA1/2/118/119 stock solutions were treated with DNase Ⅰ and RNase A(Thermo Scientific, USA)to a final concentration of 1 μg/mL, and incubated overnight at 37 ℃. Then, samples were incubated at 80 ℃ for 15 min to deactivate DNase Ⅰ. Lysis buffer(final concentrations 0.5% sodium dodecyl sulfate, 20 mmol/L EDTA, 50 μg/mL proteinase K)was subsequently added to the samples and incubated at 56 ℃ for 1 h. For DNA extraction, an equal volume of phenol was added, mixed vigorously and the mixture was centrifuged at 10, 000 × g for 10 min. The aqueous layer was removed to a fresh tube containing an equal volume of phenol-chloroform-isoamyl alcohol(25:24:1) and further centrifuged at 10, 000 × g for 10 min. The resulting aqueous layer was collected and mixed with 400 μL isopropanol, stored at -70 ℃ for > 1 h, centrifuged at 4 ℃ for 20 min at 12, 000 × g, and the final DNA pellet was washed in 1 mL 75% ethanol then centrifuged for 10 min at 10, 000 × g. The DNA pellet was then air dried at room temperature for 10 min, resuspended in 30 μL deionized water, and stored at -20 ℃ until further use.
-
Genome sequencing was performed using the semiconductor sequencer in the Life Technologies Ion Torrent Personal Genome Machine(PGM)Ion Torrent Sequencer(IonTorrent, Thermo Fisher Scientific, USA). Short read(~300 bp)data were assembled using the de novo assembly algorithm Newbler Version 2.9 with default parameters. Gene annotation was accomplished using online Tool RAST(Aziz et al., 2008; Overbeek et al., 2014; Brettin et al., 2015). The complete sequences of IME-SA1(accession number: KP687431), IME-SA2(accession number: KP687432), IME-SA118(accession number: KR902361) and IME-SA119(accession number: KR908644) were deposited in GenBank.
-
Phylogenetic trees were constructed based on 23 whole phage genome alignments. Using our four newly sequenced phages as reference, a total of 19 similar phages with query cover > 88% and identity > 97% were used for phage phylogeny reconstruction. Multiple whole genomes were aligned using Mauve V 2.3.1. MEGA6 was used to reconstruct an approximation of a maximum likelihood tree.
-
Bacteriophage IME-SA2 was centrifuged by CsCl density gradient and dialyzed in SM suspension over-night on a Transference Decoloring Shaker. Then the IME-SA2 titer was assessed by using the double layer agar technique according to methods described previously(Adams, 1959). Subsequently, the sample was negatively stained with 2%(w/v)phosphotungstate. Images were obtained using a transmission electron microscope(JEM-1200EX, Japan Electron Optics Laboratory Co., Japan)at an acceleration voltage of 100 kV.
-
Considering the four sequenced phages have variable regions directly before their terminal sequences, we designed the following primers based on the up-and downstream regions of the terminal sequence: P1(5′-GCA CAGCTAATTACAGATTACCATG-3′) and P2(5′-CC GCTTCAGCATAGATTAACAGTAG-3′). Terminal sequences of IME-SA1/2 were amplified using these primers in a PCR reaction and cloned using standard protocols. Clones containing PCR products of varying lengths were chosen for subsequent analysis.
-
To detect whether the variable region changes when an isolated MRSA phage infects different species in the genus Staphylococcus, we used IME-SA1 to infect S. aureus, S. epidermidis, and S. haemolyticus. IME-SA1 was first cultured in S. aureus and centrifuged at 10, 000 rpm for 10 min to collect the phage. The supernatant was then mixed with S. aureus and plated to acquire a single phage plaque, which was then added to 5 mL logarithmic phase culture of S. aureus, S. epidermidis, and S. haemolyticus, respectively, and allowed to grow for a further 6 h. Each culture was used as the source of respective genomic DNA to amplify the variable region using primers designed by Vector NTI(primer1: 5'-GGAGTTGTTCCCTTGTTAAC-3', primer2: 5'-CCATAGATTAAGACCGCTTC-3'). Double-distilled H2O was used as the negative control without DNA. The thermal cycling profile included an initial denaturation at 95 ℃ for 5 min, followed by 35 cycles of 94 ℃ for 30 s, 56 ℃ for 30 s, and 72 ℃ for 1 min, with an additional extension step at 72 ℃ for 7 min. PCR products were separated on a 1% agarose gel and directly sequenced using the Sanger sequencing technique by the Sangon Company(Shanghai, China).
Termini analysis theory
Phage isolation, concentration and extraction
High-throughput genome sequencing
Estimating genealogies
Transmission electron microscopy
Primers for terminal sequence analysis of IME-SA1 and IME-SA2
IME-SA1 infection of three Staphylococcus species
-
We assembled the HTS reads of IME-SA1/2/118/119 using Newbler(version 2.9, Roche). The genome size of IME-SA1 is 140, 218 bp and the reads mapping percentage was > 93%(Table 1); the genome size of IME-SA2 is 140, 906 bp and the read match was > 97%; the genome size of IME-SA118 is 139, 750 bp and the read match was > 64%(other reads belonged to S. aureus); and the genome size of IME-SA119 is 141, 026 bp and the read match was > 97%.
Bacteriophage Length (bp) # Matched Reads # Total Reads Match Percentage Min Coverage Max Coverage IME-SA1 140, 218 2, 251, 761 2, 405, 205 93.62% 676 5, 516 IME-SA2 140, 906 296, 039 288, 122 97.32% 194 1, 762 IME-SA118 139, 750 91, 643 142, 123 64.48% 36 529 IME-SA119 141, 026 2, 571, 317 2, 642, 206 97.32% 107 88, 881 Table 1. Summary of IME-SA1/2/118/1192 genome sequencing and assembly.
-
All the HTS reads from IME-SA1/2/118/119 were analyzed and ranked in descending order of HTS reads occurrence frequencies. As Figure 1 shows, IME-SA1/2/118/119 HTS read data all have one significant high-occurrence frequency read beginning with GGAATTCTTTTACCTCTCTC. The HTS data for the four phages share similar sequence occurrence frequency curves. More than 99% of reads have occurrence frequencies less than 299(IME-SA1), 134(IME-SA2), 21(IME-SA118) and 5183(IME-SA119)(Figure 2). Based on the termini analysis theory(Zhang et al., 2015), the reads with the highest occurrence frequencies represent the termini of IME-SA1/2/118/119. The ratios of the occurrence frequency of the terminal reads to general reads are 184(IME-SA1), 650(IME-SA2), 85(IME-SA118) and 3699(IME-SA119)(Table 2). Based on the phenomenon that IME-SA1/2/118/119 had only one significant high-occurrence frequency read, we hypothesize that Twortlikevirus S. aureus phages have conserved 5' terminus while the 3' terminus is variable.
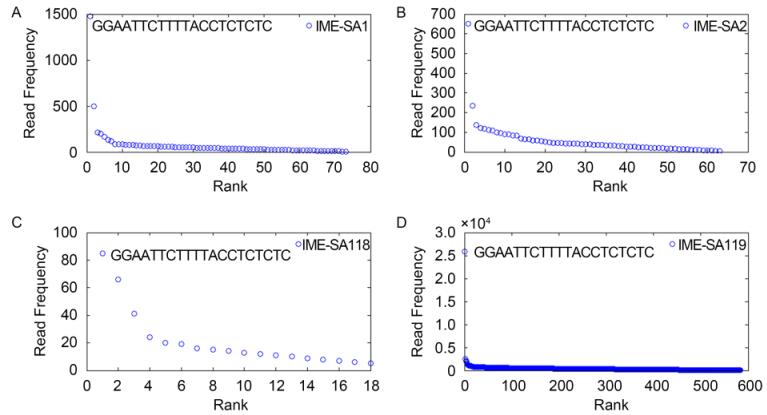
Figure 1. Read occurrence distribution. Circles indicate the initial 20 bases of HTS reads of IME-SA1 (A), IME-SA2 (B), IME-SA118 (C) and IME-SA119 samples (D). The rank in the x-axis refers to the relative frequency of each read.

Figure 2. Occurrence rates of numbers of reads. Bars indicate the initial 20 bases of HTS reads of IME-SA1 (A), IME-SA2 (B), IME-SA118 (C) and IME-SA119 samples (D). The number on the x-axis represents the frequency from the last number to the next number (e.g. 593 represent the frequency between 299 and 593).
Bacteriophage Strand Ave. Freq. Ter. Freq. Occurrence Frequency Ratio Terminal Sequence IME-SA1 Positive 8 1473 184 GGAATTCTTTTACCTCTCTC IME-SA2 Positive 1 650 650 GGAATTCTTTTACCTCTCTC IME-SA118 Positive 1 85 85 GGAATTCTTTTACCTCTCTC IME-SA119 Positive 7 25894 3699 GGAATTCTTTTACCTCTCTC Note: Ave. Freq. – Average Frequency; Ter. Freq. – Terminal Frequency. Table 2. Occurrence frequency of IME-SA1/2/118/119 terminal sequences.
-
Using the complete genomes of IME-SA1/2/118/119, a phylogenetic tree was constructed with these four phages and 19 other similar phages, using MEGA6 software for Windows(Figure 3). IME-SA1/2/118/119 grouped with Twortlikevirus S. phage G1 species. According to the International Committee on Taxonomy of Viruses(ICTV)Virus Taxonomy 2014 release(ICTV, 2014), the genus Twortlikevirus includes two species, Staphylococcus phage G1 and Staphylococcus phage K. The two species share similar terminal features. Figure 4 shows that all phages of the two species have conserved termini.
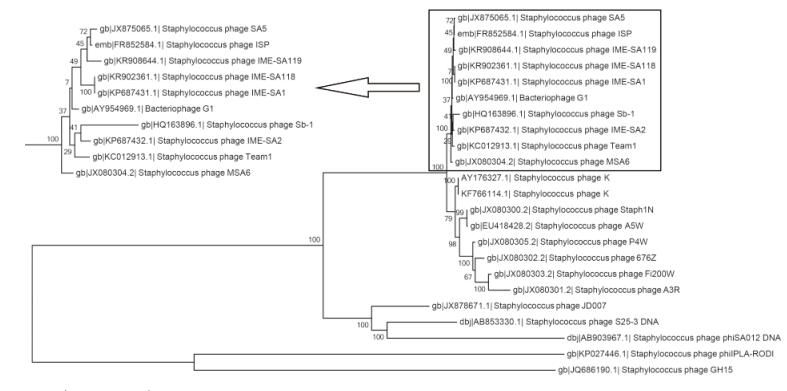
Figure 3. Neighbor-joining tree analysis based on the alignment of single nucleotide polymorphisms (SNPs) from Staphylococcus phage sequences available at NCBI. The numbers at nodes indicate bootstrap values
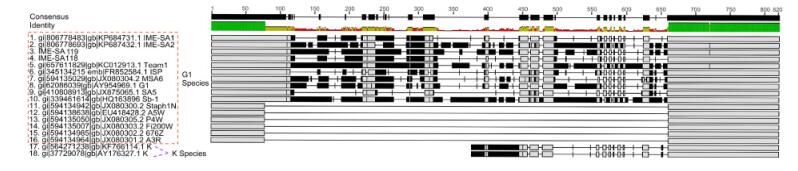
Figure 4. Comparison of the variable region in Twortlikevirus S. phages. The phages of species G1 are highlighted using the red dashed rectangle, while the K species are phages No. 17 and 18. The green bars represent the identical sequences in all sequences, while black bars show the identical sequence in some of the sequences. Numbers on the black bars illustrate the length of consensus sequence.
-
IME-SA1/2/118/119 genome analysis illustrated that the four phages have similar complete genome lengths(Table 1), all belong to G1 species(Figure 3), and that they share identical terminal sequences(Table 2) and similar terminal features(Figure 4). Thus IME-SA1/ 2/118/119 have similar characteristics. We conducted transmission electron microscopy of IME-SA2 to reveal their morphology. IME-SA2 had an isometric head(diameter about 60 nm) and a non-contractile tail(length about 200 nm)(Figure 5). According to ICTV guide-lines, IME-SA1/2/118/119 were classified as members of the Siphoviridae family, order Caudovirales.

Figure 5. Morphology of phage IME-SA2 by transmission electron microscopy.
-
From sequence analysis, we identified a variable region with a specific pattern just before the conserved terminal sequence of Twortlikevirus S. G1 species. The pattern is shown in formula(1).
*: repeats multiple times.
The details of the variable regions are shown in Supplementary Figure S1. As Supplementary Figure S1 shows, variable regions are found in all Twortlikevirus S. G1 species phages, but in neither of the Twortlikevirus S. K species. The TAC subunit repeats occur in variable numbers.
Subsequently, we cloned the phages IME-SA1 and IME-SA2, based on a previously described protocol(Green and Sambrook, 2012). Then, we chose different clones of IME-SA1 and IME-SA2 to perform terminal sequence PCR. Among the 30 sequenced clones of IME-SA1, four(IME-SA1-15, IME-SA1-18, IME-SA1-23, and IME-SA1-26) did not have the consensus IME-SA1 standard terminal sequence(Figure 6A). Similarly, among the 10 sequenced clones of IME-SA2, three(IME-SA2-6, IME-SA2-8, and IME-SA2-9) varied from the IME-SA2 standard sequence(Figure 6B).

Figure 6. PCR results of variable terminal regions in different phage clones. (A, B) Different terminal length of phage clones IME-SA1, IME-SA2. (C–E) Different terminal length of parents and offspring phage clones IME-SA1-23, IME-SA1-26, and IME-SA1-15 during successive generations. "M" represents the marker.
-
To further analyze the IME-SA1 clones that differed from the consensus IME-SA1 sequence, we cultured the respective IME-SA1 clones with the longest, average, and shortest variable sequence and then amplified their termini from the respective genomic DNA isolated from various generations using PCR. The clone with the longest terminus(IME-SA1-23) had variable terminal lengths in successive generations(Figure 6C). However, the clone with the shortest terminus(IME-SA1-26) and that with the average length terminus(IME-SA1-15) maintained the same terminal length as themselves during successive generations(Figure 6D, 6E).
-
Sequencing the variable region of four IME-SA1 and three IME-SA2 clones showed that subunit TAAGTA CCTTTGTTATG((TAC)*TAT)*(TAC)* is repeated three times in IME-SA1-15, six times in IME-SA1-18, eight times in IME-SA1-23, and twice in IME-SA1-26, with the TAC sub-subunit repeated variously(Supplementary Figure S2). Similarly, IME-SA2-6 has five repeats, IME-SA2-8 has six repeats, and IME-SA2-9 has three repeats of the subunit TAAGTACCTTTGTTATG((TAC)*TAT)*(TAC)*(Supplementary Figure S2).
Considering that IME-SA1-26 has the shortest variable region, we analyzed its HTS sequence. Among the HTS reads from IME-SA1-26, the upstream and downstream sequences of the variable region were consistent with each other while the variable regions were different from each other(51 HTS reads of IME-SA1-26 are listed in Supplementary Figure S3).
-
A comparison of the variable regions in IME-SA1 clones after infecting S. aureus, S. epidermidis, and S. haemolyticus shows no change occurred in the variable region(Data not shown).
High-throughput sequencing(HTS)analysis
Statistics of high-occurrence frequency reads in IME-SA
Phylogenetic analysis
Electron microscopy
Termini and variable region analysis
Analysis of the variable clones of IME-SA1
Sequencing IME-SA1 and IME-SA2 with variable terminal sequence lengths
Phage IME-SA1 infected three Staphylococcus species
-
In this study, the genomic DNA and termini of four newly isolated MRSA-virulent S. aureus phages, IME-SA1, IME-SA2, IME-SA118 and IME-SA119, were identified using our previously proposed termini analysis theory. Phylogeny analysis of the IME-SA1/2/118/119 HTS data shows that these phages belong to species Twortlikevirus S. phage G1. Termini analysis indicates that every Twortlikevirus S. phage has conserved 5' terminus while 3' terminus is variable. More interestingly, we discovered that a variable region exists near the conserved terminus of the Twortlikevirus S. phage G1 species. The variable region follows a pattern, as shown in formula(1). Further molecular biological experiments, including IME-SA1/2 PCR sequencing and infection of various Staphylococcus species supported our novel finding. The results show that by applying our termini analysis theory, it is possible to effectively identify phage genome termini using only HTS data, without the need for further molecular biological experiments. This is the first paper to discover the conserved terminus in Twortlikevirus S. phages and the first report of a variable region existing close to the conserved terminus of Twortlikevirus S. phages. As the terminus-adjacent variable region is a common feature of all Twortlikevirus S. G1 phages, we believe that it has potential significance for Twortlikevirus S. phage replication and packaging. Further biological experiments are needed to verify our hypothesis. In conclusion, our analysis of the newly isolated MRSA-virulent Twortlikevirus S. aureus phages IME-SA1/2/118/119 will enrich our knowledge of phages active against antibiotic-resistant S. aureus, which is essential for future antibiotic-resistant S. aureus phage therapies.
-
This research was supported by grants from the China Mega-Project on Infectious Disease Prevention(No. 2013ZX10004-605, No. 2013ZX10004-607, No. 2013ZX10004-217, and No. 2011ZX10004-001), the National Hi-Tech Research and Development(863) Program of China(No. 2014AA021402, 2012AA022-003), and the National Natural Science Foundation of China(No. 81572045).
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
YT conceived and designed the experiments and critically evaluated the manuscript. XZ was responsible for data and sequence analysis, and wrote the manuscript. HK isolated and identified the phage, conducted the biological characterization experiments. XL, YY, YL, SL collected clinical bacteria and carried out experiments. GP, ZM, YL and XA conducted the sequencing experiments. QS, PS, ZZ, YH and XX helped for sequence an-alysis. All authors read and approved the final manuscript.
Supplementary figures are available on the website of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-

Figure S1. Details of the variable region in Twortlikevirus S. phages. The subunits of the subpattern are highlighted in purple, cyan and pink, while the start and end sequences of the variable region are highlighted in green and red, respectively.

Figure S2. Details of the variable region in significant IME-SA clones. The subunits of the subpattern are highlighted in purple, cyan and pink, while the start and end sequences of the variable region are highlighted in green and red, respectively.
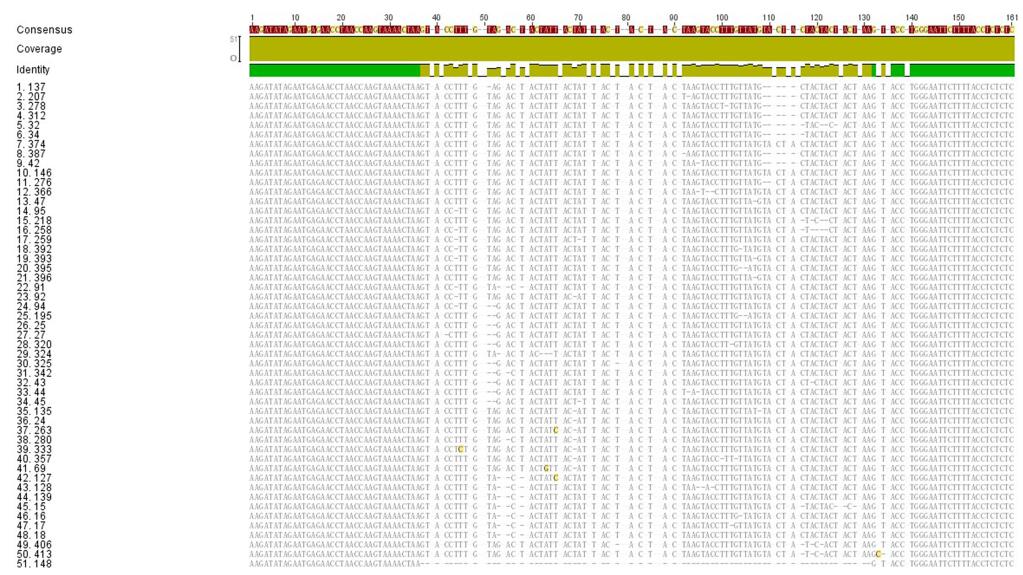
Figure S3. Comparison between the variable regions in clone IME-SA1-26.

 DownLoad:
DownLoad: