














HTML
-
Synthetic biology aims to create small artificial biological circuits and networks, which will benefit the development of new biomedical therapeutics, metabolic engineering, and energy supply (Cameron et al., 2014; Khalil and Collins, 2010). Synthetic biology originates from and progresses with technological advancements in gene manipulation (Cameron et al., 2014). Thus, it is obvious that convenient and effective methods for assembling long DNA sequences are necessary tools for this innovative research field (Merryman and Gibson, 2012).
Following the development of molecular cloning and PCR, the synthesis of DNA constructs less than 10 kb became easier. Nowadays, DNA fragments less than 20 kb can be constructed conveniently using SOEing PCR (splicing by overlap extension PCR) (Hou and Xiao, 2011; Shevchuk et al., 2004). However, a problem frequently encountered in PCR is the introduction of random errors during polymerization (Keohavong and Thilly, 1989), especially during the amplification of long and complex sequences. In the early 2000s, researchers built a circular DNA by assembling 500–800 bp segments using type IIS restriction enzymes (BsaⅠand Bbs Ⅰ) and DNA ligase (Kodumal et al., 2004). This ligation-based method had been improved to assemble as many as six DNA fragments at once. Later, another in vitro assembly method was developed by Gibson and coworkers (Gibson et al., 2008a), which eliminated the need for restriction enzyme sites within the DNA fragments. Small DNA molecules were synthesized with 20–40 bp overlapping sequences for assembly. First, exonuclease was used to chew back the 5′ ends, and DNA polymerase was then used to fill gaps in the annealed products. Finally, DNA ligase was used to covalently seal the nicks in the assembly. Based on this method, Gibson further developed a one-step, isothermal assembly method and successfully synthesized an entire 16.3-kb long mouse mitochondrial genome (Gibson et al., 2010b). Another assembly method reported by Li and Elledge (2007)was based on in vitro homologous recombination using RecA recombinase.
The construction of long DNA through homologous recombination in vivo in yeast (Stemmer et al., 1995) is an alternative method. This method was first used to ligate DNA fragments with a plasmid vector and was referred to as transformation-associated recombination (TAR) by Larionov et al. (1996). Gibson et al. applied this method to assemble large DNA molecules. Several large DNA fragments were simultaneously assembled using a yeast artificial chromosome (YAC) and yielded the Mycoplasma genitalium genome (Gibson et al., 2008a; Gibson et al., 2008b). The 1.08-Mb long Mycoplasmamycoides genome was also chemically synthesized using this method (Gibson et al., 2010a). Unlike the in vitro assembly methods, TAR-based assembly requires no enzymes and takes advantage of the homologous recombination pathway in yeast to achieve a seamless ligation of the DNA fragments. This method especially facilitates the assembly of long DNA sequences.
To improve this method, we constructed an E. coli-yeast shuttle vector for the effective assembly of long DNA fragments. The YAC regulatory and selection sequences were introduced into the BAC plasmid to generate the novel plasmid named pGF (plasmid Genome Fast), which can mediate the homologous recombination reaction, plasmid replication in yeast, and plasmid amplification in E. coli. This novel shuttle vector derived from the BAC plasmid can accommodate large DNA molecules. The assembled plasmids containing the long DNA sequences can also be easily transformed into E. coli for large-scale amplification.
Cyanophage PP is widely distributed in freshwater and may play an important role in freshwater microbial loops (Zhou et al., 2013). Using our developed shuttle vector, we assembled 10 DNA fragments from the cyanophage PP genome and produced a ~31-kb long DNA sequence. Thus, the pGF plasmid was successfully applied for the synthesis of large DNA molecules. pGF and the associated TAR assembly method will promote development in the field of synthetic biology, including viral reverse genetics and bacterial metabolic engineering (Dueber et al., 2009; Lee et al., 2013).
-
The yeast, Saccharomyces cerevisiae strain VL6–48 (ATCC), which is highly transformable and has HIS3 deletions, was used for transformation and in vivo homologous recombination. The yeast strain was cultured in st and ard rich medium containing yeast extract, peptone and dextrose (YEPD) or minimal medium CM with or without HIS. Medium supplemented with 1 mol/L D-sorbitol (Sigma Aldrich, St Louis, MO, USA) was used for spheroplast transformation. The BAC plasmid CopyControl™ pCC1BAC™ (Epicentre, Madison, WI, USA), which can accommodate large DNA fragments, was used as the vector backbone. YAC plasmid pRS313 (ATCC) was used as the PCR template to obtain the HIS3 sequence. Plasmids were electroporated into the electrocompetent cells of the E. coli strain EPI300 (Epicentre) and selected using chloramphenicol at a concentration of 25 μg/mL and ampicillin at a concentration of 50 μg/mL.
-
Small-scale isolation of supercoiled plasmids from yeast cells was performed using a TIANprep yeast plasmid DNA kit (Tiangen, Beijing, China). Briefly, the yeast cells were lysed by treatment with Lyticase. Proteins were then precipitated and eliminated, and the plasmids were further purified by affinity absorption columns. For large YACs, the circular plasmids from a large volume of yeast culture were isolated according to the protocol described by Noskov et al. (2011). Plasmid DNA from E. coli was isolated from a 5-mL overnight culture using a Plasmid Miniprep Kit (Omega Bio-tek, Doraville, GA, USA).
-
To prepare the yeast spheroplast, the cells collected from 4 mL of yeast culture grown overnight were treated with zymolyase and β-mercaptoethanol to remove the cell wall. The DNA fragments and vector were then mixed with the prepared spheroplasts as described previously (Kouprina and Larionov, 2008). Positive yeast colonies were selected on histidine-deficient (−His) plates after incubation at 30 ℃ for 2–3 days.
-
The purified plasmids were added into 100 μL of competent E. coli cells. After incubation on ice for 5 min, the cells were gently mixed and transferred into a 0.2 cm pre-chilled electroporation cuvette and pulsed with 2.5 kV, 25 μF, 200 Ω. The contents of the cuvette were then transferred into an Eppendorf tube, 1 mL SOB medium was added and incubated at 37 ℃ for 1 h while shaking at 225 rpm. All the recovered cells were then plated on LB-kanamycin plates.
-
To construct a shuttle plasmid that can replicate both in yeast and in E. coli cells, the yeast centromere sequence (CEN) and autonomously replicating sequence (ARS) were inserted into the BAC plasmid pCC1BAC, which formed the backbone. The yeast HIS3 gene was also inserted to work as a selectable marker in yeast.
The HIS3 selection marker was PCR amplified with specific primers using plasmid pRS313 as the template. The CEN6-ARS4 sequence was manually synthesized by Sangon Biotech Co. Ltd., based on the GenBank plasmid sequence with the accession number U03439. The HIS3 and CEN6-ARS4 sequences were then ligated and amplified by overlapping extension PCR. For the first step, 25 μL of PCR mixture containing 2.5 μL 10× KOD-Plus buffer, 2.5 μL 2 mmol/L dNTPs, 1.25 μL 25 mmol/L MgSO4, 0.5 μL KOD-Plus polymerase, 1.5 μL HIS3, and 1.5 μL CEN6-ARS4 templates was used without primers. The PCR conditions were as follows: 94 ℃ for 2 min, followed by 35 cycles of 94 ℃ for 15 s, 55 ℃ for 25 s, and 68 ℃ for 2 min, and a final incubation at 68 ℃ for 5 min. For the second round of PCR, the products obtained from the first round were used as the template and the primer pairs were designed to contain 5′ end overlapping sequences with ends of Afe I-linearized pCC1BAC. The PCR mixture (25 μL) contained 2.5 μL 10× KOD-Plus buffer, 2.5 μL 2 mmol/L dNTPs, 1.25 μL 25 mmol/L MgSO4, 0.5 μL KOD-Plus polymerase, 1 μL template, and 1 μL of each primer. The PCR conditions were as follows: 94 ℃ for 2 min, followed by 28 cycles of 94 ℃ for 15 s, 58 ℃ for 25 s, and 68 ℃ for 2 min, and a final incubation at 68 ℃ for 5 min. The pCC1BAC plasmid was linearized with the restriction endonuclease Afe Ⅰ, dephosphorylated using alkaline phosphatase FastAP (Thermofisher), purified, and then recovered using the E.Z.N.A. Gel Extraction Kit (Omega).
The CEN6-ARS4-HIS3 sequence and the linearized pCC1BAC plasmid were co-transformed into yeast VL6–48 spheroplasts. The positive homologous recombinants were selected on −His plates after incubation at 30 ℃ for 2–3 days. After streaking in selective media, colonies were cultured in 5 mL −His medium and the plasmids were mini-prepared. The constructed novel shuttle plasmid was verified by sequencing and named as pGF (plasmid Genome Fast).
-
The shuttle plasmid pGF was then used for the preparation of the assembly vector. This vector was PCR amplified using primers containing an approximate 20 bp overlap with ends of the BamH Ⅰ-linearized vector and a 40–60 bp overlap with the target DNA fragments. A rare-cutting restriction site NotⅠ(5'-GCGGCCGC-3') was additionally added in the middle of the primers for the easy release of the assembled products from the circular plasmids.
The DNA fragments used for the assembly were amplified by PCR using the cyanophage PP genome as the template (Zhou et al., 2013). Each assembly unit of the DNA fragments (A1, A2, A3, A4, B1, B2, B3, B4 and C1 of ~3 kb each and C2 of ~4 kb) was amplified with specific primers containing 40–60 bp overlapping sequences with the adjacent fragments (as listed in Supplementary Table S1). The PCR products were then separated using 0.6% agarose gel electrophoresis and purified using the E.Z.N.A. Gel Extraction Kit (Omega).
The DNA fragments and pGF vector were co-transformed into yeast VL6–48 spheroplasts to achieve assembly of the long DNA sequence. The positive colonies were selected on −His plates after incubation at 30 ℃ for 2–3 days.
-
Since the rare-cutting restriction site NotⅠwas introduced in the linearized pGF shuttle plasmid, the assembled intermediates or the full 31-kb DNA sequence could be released by NotⅠdigestion. EcoR Ⅰ, which also exist as the unique restriction site in pGF and the assembly products, could be used to linearize the circular plasmid for determining the length of the synthetic DNA sequences.
The assembled plasmids were isolated from the yeasts or E. coli cells as described before. PCR analyses were performed using the 2× Taq PCR MasterMix kit with the specific pairs of primers (as listed in the Supplementary material) designed to amplify all the initial DNA fragments or the sequences across the neighboring DNA fragments. The PCR mixture (10 μL) contained 1 μL extracted DNA, 1 μL each of the forward and reverse primers, and 5 μL 2× Taq PCR mixture. The PCR conditions were as follows: 94 ℃ for 2 min, followed by 32 cycles of 98 ℃ for 15 s, 58 ℃ for 30 s, and 72 ℃ for 1 min, and a final incubation at 72 ℃ for 5 min. The PCR products were loaded on 1% agarose gels and the gels were run at 100 V for 30 min.
Strains and media
Isolation of plasmid DNA from yeast or E. coli
Co-transformation of the DNA fragments into yeast
Electroporation of E. coli
Construction of the E. coli-yeast shuttle vector, pGF
Preparation of the assembly vector and cyanophage PP fragments
Restriction digestion and PCR analysis of the assembled long DNA sequences
-
As shown in Figure 1, the shuttle plasmid was constructed based on the BAC plasmid pCC1BAC and YAC plasmid pRS313. pCC1BAC normally replicates as a single copy under the regulation of the F-factor, and SopA, B, and C are all important for plasmid partitioning during cell division in E. coli. When an inducer is added, the number of plasmid copies could be increased to 10–20 in the EPI300 strain under the control of oriV. Besides, the phage COS sequence in the plasmid enables it to accommodate large-size DNA sequences. The YAC plasmid pRS313 contains CEN6 and the ARS associated with HIS3 elements. The yeast centromere CEN6 and the autonomous replicating sequence ARS enable the plasmid to replicate in yeast cells, while the HIS3 is a selectable marker for positive colony screening. The yeast regulatory component CEN6-ARS4 and the HIS3 sequences were introduced into the appropriate sites of pCC1BAC. This novel synthetic plasmid can mediate the homologous recombination of DNA fragments and stably maintain the assembled plasmids in the yeast. Further, the assembled plasmids purified from the yeast cells could be transformed into E. coli for amplification.
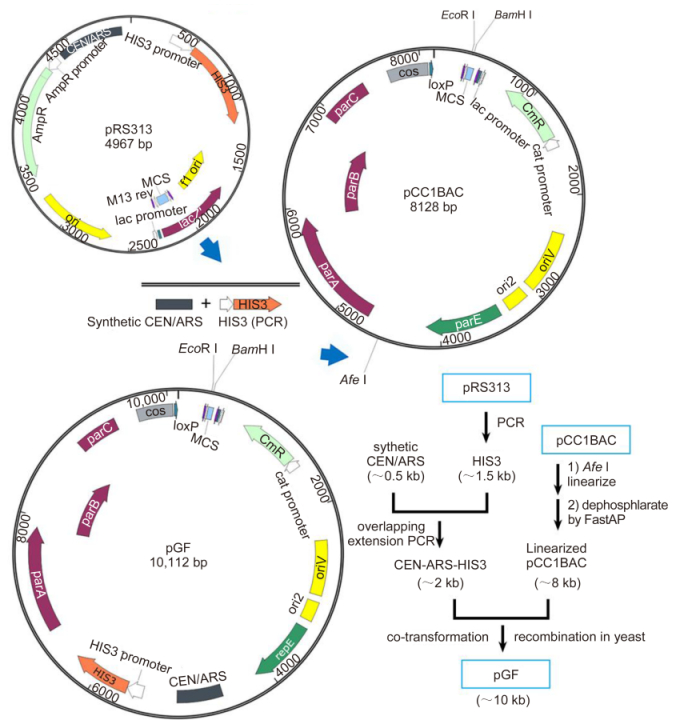
Figure 1. The construction of pGF shuttle plasmid used for in vivo TAR assembly of DNA fragments. The BAC plasmid pCC1BAC used as the backbone allows the replication of the assembled DNA in E. coli for amplification. CEN6-ARS4 and HIS3 sequences introduced at the AfeⅠsites of pCC1BAC allow the propagation of plasmids and positive colony selection in yeast.
To construct the shuttle plasmid pGF described above, the CEN6-ARS4 and HIS3 sequences were inserted into the AfeⅠsite of the BAC plasmid pCC1BAC. Specifically, the HIS3 sequence (~1.5 kb) was PCR amplified from the YAC plasmid pRS313, and the CEN6-ARS4 sequence (~0.5 kb) was obtained by DNA synthesis based on the GenBank sequence with the accession number U03439. These two sequences were tandemly linked by overlapping extension PCR to generate the CEN6-ARS4-HIS3 sequence (~2 kb). Then, the CEN6-ARS4-HIS3 sequence was co-transformed with the Afe Ⅰ-linearized pCC1BAC vector (~8 kb) into yeast and ligated by TAR homologous recombination. The positive colonies were selected and plasmids were isolated and sequenced. The successful assembly of the pGF vector (~10 kb) by TAR cloning indicated that this in vivo homologous recombination system in yeast could effectively ligate the DNA fragments into the vector with the 5′ end overlapping sequences.
-
Figure 2 shows the flowchart for the assembly of 10 cyanophage PP DNA fragments into a ~31-kb DNA molecule. In the first step, the DNA fragments of Part A (A1, A2, A3 and A4, each of ~3 kb), Part B (B1, B2, B3 and B4, each of ~3 kb), or Part C (C1 of ~3 kb and C2 of ~4 kb) with the pGF vector were co-transformed into yeast. The neighboring fragments with overlapping ends could be ligated together by homologous recombination to generate pGF-Part A, Part B, or Part C, respectively. These plasmids were then digested by NotⅠso that the three intermediates Part A, Part B, and Part C could be released and recovered by agarose gel separation and extraction. Next, the assembled intermediates Part A (~12 kb), Part B (~12 kb) and Part C (~7 kb) with the vector pGF (~10 kb) were co-transformed into yeast cells and further assembled to form the 31-kb long DNA sequence.

Figure 2. Scheme for the assembly of the ~31-kb DNA sequence. In the first stage of assembly, four DNA fragments of Part A and Part B and two DNA fragments of Part C were ligated together to generate the intermediates pGF-Part A, Part B, and Part C. In the next step, the assembled Part A, Part B, and Part C were released by digestion by the endonuclease NotⅠ and recovered. The three intermediates together with pGF were then co-transformed into yeast cells to generate the complete ~31-kb PartABC integrated sequence in the pGF vector.
According to the homologous recombination strategy, the pGF vector and the cyanophage PP DNA fragments should be prepared by PCR amplification to add the specific overlapping ends. The 5′ ends of the primers were designed to contain 20–40 bp extension sequences homologous to the neighboring fragments. A rare-cutting restriction site Not Ⅰ (5'-GCGGCCGC-3') adjacent to the overlaps needed to be added to the primers for the amplification of the pGF vector for the convenient release of the assembly products. The products of PCR amplification of the pGF vector performed using high fidelity polymerase were exhibited as the main electrophoresis bands approximately 10 kb in size (Figure 3A). The Part A (A1, A2, A3 and A4, each of ~3 kb), Part B (B1, B2, B3 and B4 each of ~3 kb), and Part C (C1 of ~3 kb and C2 of ~4 kb) DNA fragments were also PCR amplified and purified using the Gel Extraction Kit (Figure 3B). Alternatively, these ~3-kb long DNA fragments also could be obtained by chemical synthesis as described before (Hou and Xiao, 2011).
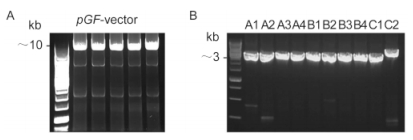
Figure 3. Preparation of the pGF vector and cyanophage PP DNA fragments. (A) The ~10-kb pGF vector was amplified using PCR and the homologous 5′ ends bracketing the inserted DNA fragments were inserted. (B) The DNA fragments A1-C2 were amplified using PCR from the cyanophage PP genome. Similarly, the 5′ end overlapping sequences were introduced using primer pairs.
The three assembled DNA intermediates together with the pGF vector were then co-transformed into yeast cells to generate pGF-Part ABC. As indicated in Figure. 4B, the EcoR Ⅰ-linearized pGF-Part ABC was about 41 kb in size (lane 2), as determined using the high molecular weight marker, which was in accordance with the full length of the target product. As mentioned above, the plasmid pGF-Part ABC could release the inserted assembled long DNA sequence upon treatment with Not Ⅰ. Lane 3 in Figure 4B shows that two b and s of ~10 kb and ~31 kb were separated on a 0.6% agarose gel, which corresponded to the free pGF vector and the assembled Part ABC, respectively.
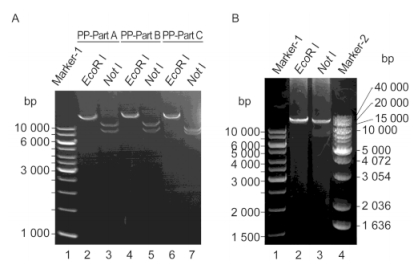
Figure 4. Restriction digestion analysis of the assembled DNA sequences. (A) Verification of the assembled intermediates of Part A, Part B, and Part C. Yeast colonies containing positive assemblies were verified by digestion with the endonucleases EcoRⅠ(lanes 2, 4 and 6) and NotⅠ(lanes 3, 5 and 7). A molecular weight marker was loaded in lane 1. (B) Analysis of the ~41-kb assembled pGF-PartABC by digestion with the endonucleases EcoRⅠ(lane 2) and NotⅠ(lane 3). Standard and high molecular weight markers were loaded in lane 1 and lane 4, respectively.
To further verify the positive colonies, the assembled plasmids were extracted and analyzed by PCR. As shown in Figure 5A, the PCR performed using the primer pairs of each DNA fragment all generated products with target sizes (A1, A2, A3, A4, B1, B2, B3, B4 and C1, each of ~3 kb and C2 of ~4 kb), which indicated that all the 10 cyanophage PP DNA fragments were present in pGF-Part ABC. Next, specific primer pairs (FV/RA1, FA1/RA2, FA2/RA3 and so on; Figure 5B) were designed to amplify the sequences across the two adjacent DNA fragments (as listed in Supplementary Table S2). The correct order of the assembly was verified by PCR amplification using these primers. The result showed that all the PCR products separated on the agarose gel exhibited molecular sizes consistent with those of the designed target products (indicated below the amplified fragments in Figure 5B). This result further confirmed the fact that the 10 cyanophage PP DNA fragments were assembled in the desired order and formed the 31-kb long DNA sequence.

Figure 5. PCR analysis of the assembled ~41-kb long DNA sequence. (A) Amplicons of all the 10 cyanophage PP DNA fragments were present in the complete assembled product pGF-PartABC. (B) Amplification of the DNA sequences across the adjacent fragments. To ensure the correct order of the assembled DNA fragments, specific primers located on the inner side of each fragment were designed. The forward primers of the pGF vector (FV) and the reverse primer of A1 (RA1) were used in pairs to amplify the sequences across the vector and A1. Similarly, FA1/RA2, FA2/RA3, and so on were used in pairs to amplify the sequences across two adjacent fragments. The precise sizes of the target PCR products are marked.
Construction of the shuttle plasmid pGF
Assembly of the 31-kb long DNA from the cyanophage PP genome
-
The assembly of long DNA molecules requires a more accurate and effective scheme rather than a routine ligation-based cloning method. Although PCR enzymes with high fidelity have been developed, the random errors introduced during the amplification process are an obvious limitation of long sequence PCR. The complexity of different DNA templates influences the accuracy of PCR, especially in the case of amplification of long DNA sequences (Kumar and Kaur, 2014; Varadaraj and Skinner, 1994). The in vitro Gibson assembly method can ligate several DNA fragments to produce large DNA sequences by using single-str and DNA 3′ overhangs. However, this method needs exonucleases to chew back from the DNA 5′ end, DNA polymerase to fill the gaps, and DNA ligase to remove nicks, which increases the cost of using this method. In addition, the concentrations of the DNA fragments and enzymes must be regulated accurately in order to achieve successful assembly. In addition, the yield of positive colonies may decrease when assembling more than 5 DNA fragments at once.
The shuttle plasmid pGF and in vivo TAR assembly method described here provides an alternative way to combine DNA fragments to form large DNA sequences. The shuttle plasmid pGF containing the YAC regulatory elements could be ligated with several DNA fragments with overlapping ends through homologous recombination in yeast. Moreover, the assembled products could be amplified in E. coli.
Using the yeast recombination system, the appropriate DNA fragments with 5′ end overlapping sequences could be assembled in the yeast cells. Notably, the 5′ overlapping sequences could be introduced into the DNA fragments by PCR using synthetic specific primers, thus eliminating the need for the chewing back step by exonuclease, which was required in the Gibson model. The homologous recombination-based TAR method is very helpful especially for the assembly of long DNA sequences. Using this method, the assembly and the modification of a viral genome from small DNA fragments could be conveniently achieved. The fidelity of the assembled large DNA molecule was only associated with the fidelity of the assembly units (relatively small DNA fragments). The sizes of the small DNA fragments used for the assembly were designed to be below 5 kb, so that the fidelity of the synthetic large DNA molecule could be guaranteed. Alternatively, DNA fragments less than 5 kb in size can also be easily obtained by using the DNA synthesizer.
Genomes of living organisms will always contain some gene regulatory elements such as long terminal repeat (LTR) sequences. It should be noted that these sequences always have a high G + C content and may form complex secondary structures, thus influencing effective assembly. To ensure the fidelity of such DNA fragments, a specific high-fidelity polymerases (such as Q5 High-Fidelity DNA polymerases from NEB) designed to amplify the high G + C template should be used, and DMSO and other reagents should be added at appropriate concentrations to lower the Tm value. In addition, the sequences that readily form complex secondary structures should be arranged to be in the middle of the DNA fragments, and not at the 5′ or 3′ ends. Regulatory sequences may also sometimes be toxic for E. coli. The recombination-based assembly in yeast could circumvent the toxicity to E. coli, but the assembled products could be prepared only from large-scale culture of yeast cells in this case.
In conclusion, the present work provided a novel synthetic E. coli-yeast shuttle plasmid pGF and a TAR-based assembly method for the construction of large DNA sequences. Using this method, the reconstruction of a mini genome of various viruses from the relatively small torque teno virus (TTV) and adeno-associated virus (AAV) to the large baculovirus like autographa californica multicapsid nucleopolyhedrovirus (AcMNPV) could be achieved efficiently. The novel synthetic shuttle plasmid pGF and the associated DNA assembly method will not only aid in the design of artificial biological systems like in bacterial metabolic engineering, but will also accelerate the progress of synthetic virology including mini-genome construction and vaccine research and development against new recombination viruses.
-
We thank Dr. Zhihong Hu, Dr. Zhengli Shi, and Dr. Fei Deng of the Wuhan Institute of Virology, Chinese Academy of Sciences, China, for their advice, encouragement, and support in conducting this work. This study was supported by the 973 program, Grant No. 2012CB721102.
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
ZH and ZLW carried out experiments for the construction of the pGF vector and cyanophage PP genome assembly; ZZ wrote the manuscript and participated in some of the experiments. GFX was the corresponding author who conceived and supervised the project.
Supplementary Tables are available on the website of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-
Primer name Sequences nt Primers used for amplification of pGF vectors XAF-1 GCTCGTAGATCGTCATGGTTTGTTTCTCACCTGGTAGTTCgcggccgcgatcctctagagtcgacctg 68 XAR-1 AAGCTTAGGTACTTCTGGTAACGGTTTACGAGTGACGACAgcggccgccgggtaccgagctcgaattc 68 XBF-1 CCTCCTGGTCGTACAGCAACACCAACTAGACGACTACCGCgcggccgcgatcctctagagtcgacctg 68 XBR-1 GTGACGGAGCGTCGTAGAGAACGGTAGCTCGTGACGGTTGgcggccgccgggtaccgagctcgaattc 68 XCF-1 AAAGCGAAGACTTGCCCGAACTCCCGTCACTTGAAGACACgcggccgcgatcctctagagtcgacctg 68 XCR-1 GCTGGCCGACCTGCTCGACGCTATCGAAGACGACGCACAGgcggccgccgggtaccgagctcgaattc 68 Primers used for amplification of fragments A1-A4, B1-B4, C1-C2 AF1 GAACTACCAGGTGAGAAACAAACCATG 27 AR1 ACCATCGTTACAGGTTTTAACATCGG 26 AF2 TGACTTGGCACGGTGTACTAGGTCAC 26 AR2 GCTGCTACGTTTGTAGACCTAACCTATAAG 30 AF3 TCTGATAGCATCATCAAAGCCGTTAC 26 AR3 ACCTATGTTGTCTGGTCGGTCGTTT 25 AF4 CTTTGATGATGACAGCCGTAGGTTT 25 AR4 TGTCGTCACTCGTAAACCGTTACCAG 26 BF1 GCGGTAGTCGTCTAGTTGGTGTTG 24 BR1 CACAGTGGAACCAACAACTACTAGAAGC 28 BF2 GGTCACGTAAGCTTGAGATCACCTCT 26 BR2 GTACTTCGACAACGAAGATGTGCTACC 27 BF3 CTACTAAGTCATCTACATCCTGACCAGAAGC 31 BR3 GAAAAGCAAGCACAACGAGCTAAGC 25 BF4 ACAGCTTGTTGTCTTTTTCTTTCACGTATC 30 BR4 CAACCGTCACGAGCTACCGTTC 22 CF1 GTGTCTTCAAGTGACGGGAGTTCG 24 CR1 ATGCTTGTCGTAGACCCTGCTGTG 24 CF2 TCGTTGTCGTAACCACCTACGGTC 24 CR2 CTGTGCGTCGTCTTCGATAGCGTCGAG 27 Table S1. Primers used for PCR amplification of pGF vectors and fragments
Primer name Sequences nt FV CCCAGGCTTTACACTTTATGCTTC 24 RA1 GCAACCGACACACAGCCTCTT 21 FA1 TACACGGGTGGGGTGTTAGTACC 23 RA2 CTTCTGCTTTTGAAAGACCTCCTCT 25 FA2 CTGGGTCGTACATGATAGCTGCTG 24 RA3 TGGTCACTTACAACGTATCATCCCT 25 FA3 CTTGTTCACCTCGTCTACTAACCG 24 RA4 AGTGCTACTAACTGGGTTGTCGATG 25 FA4 GTTCTACAGAAGGACGTGGTTGTGT 25 RB1 GTAACAACAGCTACACTGTGGCTG 24 FB1 CTGTGCTCGTTGTCGTTCTACTG 23 RB2 AGAAGGGTGAGATACAACGCTTTG 24 FB2 CACAGAGTTTCCACGAAGCCTC 22 RB3 CTTCAAGTAGTGCCTTACGACAACG 25 FB3 CCTTGTCGTTTACTGACGTTACCAC 25 RB4 GTTATCAAACGTGCGTCTTACCTCA 25 FB4 TTTTCCCAGAGTTTACGCACCTC 23 RC1 TAGTTCCTATCGGAAGCGACCAT 23 FC1 GAAGTTGTACACTTTGGCGGGAG 23 RC2 CTATCACTACAACCCAGTAGCCAACA 26 FC2 GGGCAAGGCTCTTAGGGTCTTCATTC 26 RV GTGAAATACCACACAGATGCGTAAG 25 Table S2. Primers designed to amplify the sequences across the adjacent DNA fragments

 DownLoad:
DownLoad: