












HTML
-
To date, six coronaviruses infecting humans have been characterized. Infections with human coronaviruses (HCoVs)229E (Hamre & Procknow, 1966), OC43 (McIntosh et al., 1967), NL63 (van der Hoek et al., 2004), and HKU1 (Woo et al., 2005) cause relatively mild symptoms in most cases, whereas severe acute respiratory syndrome coronavirus (SARS-CoV; Drosten et al., 2003; Ksiazek et al., 2003; Kuiken et al., 2003; Peiris et al., 2003) and Middle-East respiratory syndrome coronavirus (MERS-CoV; Zaki et al., 2012) are connected with severe respiratory-tract infection and, in particular in case of MERS-CoV, acute renal failure (Eckerle et al., 2013), leading to high case-fatality rates of ~10 and ~35%, respectively. In spite of 13 years of research on SARS-CoV (Hilgenfeld & Peiris, 2013), no approved drugs or vaccines are available for the treatment or prevention of coronavirus infection (Wang et al., 2016). This is mainly due to the fact that although these emerging viruses have devastating effects on those infected, the absolute numbers of cases (~8000 for SARS, 1733 so far for MERS; (http://www.who.int)) imply that the development of specific antivirals is very likely not commercially viable. On the other hand, the global risk posed by MERS-CoV must not be underestimated. Since its discovery in September 2012, the number of MERS cases reported has been rising steadily, with some intermittent peaks connected to hospital outbreaks in Saudi Arabia (Assiri et al., 2013). Man-to-man transmission of MERS-CoV has also been impressively demonstrated by the recent outbreak of MERS in South Korea, which was traced back to a single traveller from the Arab peninsula (Butler, 2015). Therefore, it is imperative that academic laboratories help increase the preparedness against a possible MERS-CoV pandemic by characterizing antiviral drug targets and by identifying lead compounds interfering with them.
In order to successfully infect humans, a virus has to meet at least two conditions: 1), it should maintain a sufficiently correct replication of its genetic material; 2), it should inhibit the host antiviral response. The papain-like protease (PLpro) of MERS-CoV (or SARS-CoV) is involved in both of these tasks (Yang et al., 2013; Barretto et al, 2005). The PLpro is a domain located in the middle part of the largest non-structural protein, Nsp3, of MERS-CoV (or SARS-CoV). It is responsible for releasing Nsp1, Nsp2, and Nsp3 from the polyproteins 1a (pp1a) and 1ab (pp1ab), an essential step of replication (Harcourt et al., 2004). Like its SARS-CoV counterpart, the MERS-CoV PLpro also has deubiquitinating (DUB) and deISGylating activities in vivo as well as in vitro (Yang et al., 2013; Mielech et al., 2014; Lei et al., 2014; Baez-Santos et al., 2014b). K48-and K63-polyubiquitin poly (Ub) and ISG15 (interferon-stimulated gene 15)-conjugated targets are usually involved in host innate immune regulation (Liu et al., 2005; Maringer & Fernandez-Sesma, 2014). The PLpro has the ability to digest K48-and K63-linked polyUb chains and to remove ISG15 from ISG15-linked proteins (Baez-Santos et al., 2014b), thereby interrupting the signalling pathways leading to the innate immune response. Thus, the PLpro can block the activation of IFN regulatory factor 3(IRF3) (Yang et al., 2013) and subsequently the production of interferon β(IFNβ) (Mielech et al., 2014). Interestingly, MERS-CoV PLpro shows a similar cleavage rate for K48-and K63-linked polyUb chains, while the SARS-CoV enzyme prefers K48-over K63-linked chains (Baez-Santos et al., 2014b). The former enzyme degrades a polyUb chain by removing mono-Ubs, whereas the latter cleaves di-Ub units off the polyUb chain (Bekes et al., 2015).
We have reported the first crystal structure of the MERS-CoV PLpro(Lei et al., 2014). Later, two other groups also described the structure of this enzyme (Bailey-Elkin et al., 2014; Lee et al., 2015). The structure of PLpro can be divided into two parts: a ubiquitin-like (Ubl) domain and a catalytic domain with thumb, palm, and fingers subdomains. The overall fold of MERS-CoV PLpro is not only similar to that of SARS-CoV PLpro, but also to that of several human ubiquitin-specific proteases (USPs) (Hu et al., 2005). In 2014, the X-ray structure of the complex of SARS-CoV PLpro with ubiq-uitin has been reported (Chou et al., 2014; Ratia et al., 2014). Several key residues (such as Glu168 or Tyr265) of SARS-CoV PLpro that are important for ubiquitin recognition (Chou et al., 2014; Ratia et al., 2014), are not conserved in MERS-CoV PLpro. Bailey-Elkin et al.(2014) described the structure of an artificially linked, covalent complex between ubiquitin and MERS-CoV PLpro. Here, we present the crystal structure of a non-covalent complex between the two proteins and a mutational study of the interactions involved. For these studies, we used the Cys111Ser active-site variant of MERS-CoV PLpro.
-
The PLpro of MERS-CoV (strain 2c EMC/2012; GenBank no. AFV09327.1) contains 320 residues, from Gln1482 to Asp1801 of pp1a/1ab. For simplification, Gln1482 was renumbered into Gln1 here. The DNA plasmid coding for MERS-CoV PLpro was produced earlier (Lei et al., 2014).
The MERS-CoV PLpro C111S, D164E, D164A, D165E, D165A, and F269Y variants were produced using the same strategy that we described before (Lei et al., 2014). All primers for these variants are listed in Supplemental Table S1. All DNA plasmids coding for the altered PLpro were verified by sequencing.
Genes coding for wild-type (WT) MERS-CoV PLpro and for its variants were expressed and the corresponding proteins were purified according to our previous description (Lei et al., 2014).
-
The PLproof SARS-CoV (Strain TOR2; GenBank no. AY274119.3) comprises 319 amino-acid residues, corresponding to Glu1541 to Tyr1859 of pp1a/1ab. A gene coding for the SARS-CoV PLprowas amplified by PCR with the following two primers 5'-CTAGCTAGCGAGGTTAAGACTATAAAAGTGTTC-3'(forward) and 5'-CCGCTCGAGTTAATACGACACAGGCTTGATGGTTGTAG-3'(reverse). The PCR product was digested by Nhe Ⅰ and Xho Ⅰ, then was ligated into the pET-28a plasmid (Novagen). The recombinant plasmid DNA was verified by sequencing. Expression of the gene construct coding for SARS-CoV PLpro and purification of the protein were performed according to the procedure described for MERS-CoV PLpro(Lei et al., 2014).
-
Purified MERS-CoV PLpro(WT) and a variant that had the active-site Cys111 replaced by Ser (PLpro(C111S)) were both concentrated to ~24 mg/mL in 20 mmol/L Tris-HCl, 150 mmol/L NaCl, pH 8.8, 10 mmol/L β-mercaptoethanol (BME). Human ubiquitin (BostonBiochem) was dissolved to 6 mg/mL in 20 mmol/L Tris-HCl, 150 mmol/L NaCl, pH 8.8. 500 μL PLpro(WT) or PLpro(C111S) were mixed with 500 μL ubiquitin (~1:1 molar ratio) at 4 ℃ overnight. The complex of PLpro(WT)-Ub or PLpro(C111S)-Ub was purified by gel filtration (HiLoadTM 16/60 S200 column, GE Healthcare) in 20 mmol/L Tris-HCl, 150 mmol/L NaCl, pH 8.8 the next day. The final concentration of PLpro(WT)-Ub or PLpro(C111S)-Ub was ~12 mg/mL. The two complexes were crystallized using the sitting-drop method and a Phoenix crystallization robot (Art Robbins) at 18 ℃. 0.25 μL of protein and 0.25 μL of reservoir were mixed and equilibrated against 75 μL reservoir. Screening kits IndexTM, SaltRxTM, PEG RxTM 1 & 2, PEG/IonTM 1 & 2(Hampton Research), and Structure Screen 1 MD1-01, Structure Screen 2 MD1-02(Molecular Dimensions) were used. Crystals of PLpro(C111S)-Ub were observed under condition No. 9 of MD1-01, whereas no crystal of PLpro(WT)-Ub was obtained. Optimized crystals of PLpro(C111S)-Ub were subsequently obtained within two days under the condition: 22% w/v PEG 4000, 15% v/v 2-propanol, 0.1 mol/L tri-sodium citrate pH 4.8, and 10% glycerol. 2 μL of protein and 2.5 μL of reservoir were mixed to equilibrate against 500 μL reservoir.
Crystals were placed in a nitrogen-gas stream (100 K). A 3.16-Å dataset was collected at wavelength 0.91841 Å at beamline 14.2 of BESSY, Berlin (Mueller et al., 2012). The program XDS (Kabsch, 2010) was used to process the diffraction data. The space group was found to be P63, with unit-cell parameters a = b =138.14 Å, c = 57.59 Å, γ = 120°. Diffraction data statistics are shown in Table 1.
-
The structure of the MERS-CoV PLpro(C111S)-Ub complex was solved by molecular replacement using MOLREP (Vagin & Teplyakov, 2010). The program selected the MERS-CoV PLpro(Protein Data Bank (PDB) entry 4P16, Lei et al., 2014) as the first search model. Human ubiquitin (PDB entry: 1UBQ, Vijay-Kumar et al., 1987) was used as the second search model. The model of the complex was inspected and rebuilt using Coot (Emsley et al., 2010), and refined using phenix.refine (Headd et al., 2012). The refinement statistics are shown in Table 1. The final model coordinates and structure factors have been deposited in the PDB database with the code 4WUR. Figures (except for the supplemental figure) have been prepared using Pymol (Schrö-dinger; http://www.pymol.org/).

Table 1. Data collection and refinement statistics
-
All enzymatic assays were performed using a 96-well microtiter plate and the reaction buffer 20 mmol/L Tris-HCl, 150 mmol/L NaCl, pH 7.9, 2 mmol/L dithiothreitol (DTT). The fluorogenic substrates Cbz-Arg-Leu-Arg-Gly-Gly-7-amino-4-methylcoumarin (Z-RLRGG-AMC) (Bachem), Z-LRGG-AMC (BostonBiochem), and ubiq-uitin-AMC (Ub-AMC) (BostonBiochem) were used. The fluorescence of free AMC with different concentrations (5 nmol/L-2.5 μmol/L) in reaction buffer was measured to generate a calibration curve, in order to convert the change of fluorescence intensity per unit of time, Δ(AFU)/s, into the amount of hydrolyzed substrate in μmol/L/s.
The enzymatic cleavage reactions were run with an Flx800 fluorescence spectrophotometer (BioTek), to measure the increased fluorescence signal (λex: 360 nm; λem: 460 nm) resulting from AMC release. Reactions were initiated by addition of the proteases to the reaction system. The peptide-hydrolysis kinetic assays were performed with the following conditions: 1 μmol/L MERS-CoV PLpro variant (D164E, D164A, D165E, F269Y), or 10 μmol/L D165A, or 0.1 μmol/L SARS-CoV PLpro, with different concentrations (10, 20, 40, 80, 100 μmol/L) of Z-RLRGG-AMC or Z-LRGG-AMC in a final volume of 100 μL at 25 ℃. The kinetic curves for the proteases and their variants with the substrates Z-RLRGG-AMC or Z-LRGG-AMC were linear and the initial velocities also increased linearly with substrate concentration. No satura-tion was observed. Therefore, the data were fitted to the equation v/[E]tot. = kapp[S], where kapp approximates kcat/KM, as described previously (Barretto et al., 2005; Wojdyla et al., 2010).
The deubiquitinating kinetic assays were performed under the following conditions: 0.1 μmol/L MERS-CoV PLpro wild-type or its variants D164E, D164A, D165E, F269Y, or 0.5 μmol/L D165A, or 0.025 μmol/L SARS-CoV PLpro were incubated with increasing concentrations (1, 2, 4, 8 μmol/L) of Ub-AMC in a final volume of 50 μL, at 25 ℃. Although PLpro actually cleaves the isopeptide bond between the carboxyl group of the C-terminal Gly in Ub and the ε-amino group of Lys in ubiq-uitinated targets in vivo, we used the hydrolysis of Ub-AMC here to test the deubiquitinating activity in vitro. The kinetic curves of proteases and variants with the substrate Ub-AMC were hyperbolic and the initial veloci-ties were not linear over the concentration of substrate. However, saturation was still not observed within a rea-sonable time. Only when the ratio of protein to substrate was 1:1 or larger, were we able to achieve saturation within a limited time (data not shown). As the initial velocities did not increase in a strictly linear fashion with substrate concentration, application of the equation v/[E]tot. = kapp[S] to mimic kcat/KM would lead to large standard errors. We were however able to fit the data to the Michaelis-Menten equation using the GraphPad Prism program (GraphPad Software), even though satura-tion could not be reached (Supplemental Figure 1). All assays were performed in duplicates.
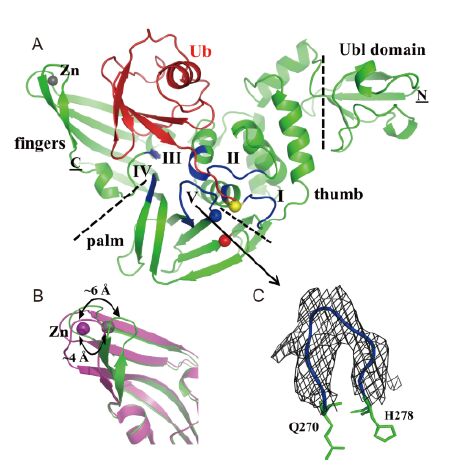
Figure 1. Structure of MERS-CoV PLpro in complex with human ubiquitin (Ub). (A) Cartoon view of the overall complex structure. PLpro is shown in green and Ub in red. The N and C termini are marked by underlined letters, and the PLpro subdomains are divided by black dashed lines. The ubiquitin-like (Ubl) domain, thumb, palm, and fingers subdomains are indicated. The catalytic triad Cys111-His278-Asp293 is shown by spheres (yellow, blue, and red; Cys was replaced by Ser in this study). The zinc atom is displayed as a gray sphere. Five PLpro regions interacting with Ub are colored dark blue and labeled with Roman numbers (Ⅰ-Ⅴ). (B) Superposition of the fingers domain of Ub-bound MERS-CoV PLpro and the substrate-free enzyme (PDB: 4P16; Lei et al., 2014). The zinc atom is shifted by ~4 Å, and the largest difference is ~6 Å. (C) The BL2 loop (blue; 271GIETAVG277) of PLpro. A 2Fo-Fc electron density (gray; 1.0 σ) is displayed. The side-chains of residues in this loop are only partly defined by electron density.
Recombinant production of MERS-CoV PLpro and its variants
Recombinant production of SARS-CoV PLpro
Crystallization of MERS-CoV PLpro with ubiquitin
Phase determination, model building and refinement
Kinetic assays of purified PLpros
-
The substrate-binding site of MERS-CoV PLpro features significant differences from those of the corresponding SARS-CoV enzyme and human ubiquitin-specific proteases (USPs, such as, USP14) (Hu et al., 2005; Chou et al., 2014; Ratia et al., 2014). It is therefore of interest to determine the crystal structure of the complex between MERS-CoV PLpro and its substrate, human ubiquitin. Hence, we crystallized the ubiquitin (Ub) complex of a MERS-CoV PLpro variant that had the active-site Cys111 replaced by serine (C111S) and determined the structure at 3.16 Å(Figure 1A).
There is one PLpro(C111S)-Ub complex per asymmetric unit. Using the PDBePISA server (Krissinel & Henrick, 2007), the total interface region of MERS-CoV PLpro(C111S)-Ub was determined as 813 Å2, close to the 915 Å2 interface of SARS-CoV PLpro-Ubal (ubiquitin aldehyde; PDB entry: 4MM3, Ratia et al., 2014) and the 999 Å2 of SARS-CoV PLpro(C112S)-Ub (PDB: 4M0W, Chou et al., 2014), but less than the 1503 Å2 observed for USP14-Ubal (PDB: 2AYO, Hu et al., 2005). The overall structure of MERS-CoV PLpro(C111S) is very similar to that of the substrate-free PLpro(Lei et al., 2014), with a root-mean-square deviation (RMSD) of 0.91 Å for corresponding Cα atoms between these two structures. Nevertheless, two differences are immediately visible: 1), the zinc-finger motif has moved and closed in onto the Ub, compared to the free PLpro. The zinc ion position has shifted by about 4 Å and the largest deviation between the two structures is ~6 Å for Cys228 of the zinc-finger region (Figure 1B); 2) the mobile loop 271GIETAVG277, also named "BL2 loop", is defined by clear main-chain electron density (Figure 1C). This loop is disordered in substrate-free PLpro(Lei et al., 2014; Bailey-Elkin et al., 2014). Compared to MERS-CoV PLpro, the BL2 loop 267GNYQCG272 is shorter by one residue in SARS-CoV PLpro.
At the time when we deposited in the PDB the coordinates for the crystal structure of the non-covalent complex MERS-CoV (C111S)-Ub (PDB: 4WUR), Bailey-Elkin et al.(2014)described two crystal structures for a covalent complex MERS-CoV PLpro-Ub, in which an alkyl bromide group introduced at the C-terminus of Ub had formed a thioether with the active-site Cys111 of MERS-CoV PLpro. These structures were in space groups P6522 and P63 and were named "closed" and "open" PLpro-Ub complexes (PDB: 4RF0 and 4RF1), respectively (Bailey-Elkin et al., 2014). Even though the zinc-finger motif of PLpro in the former complex (space group P6522) is closer to the Ub than in the P63 complex, both the "closed" and "open" complex show almost the same interactions between PLpro and Ub (Bailey-Elkin et al., 2014). The differences may be caused by crystal packing. Our non-covalent complex is similar to the "open" form of the covalent PLpro-Ub complex (RMSD = 0.58 Å for the PLpro and 0.85 Å for Ub, based on all Cα atoms).
All parts of the PLpro except for the ubiquitin-like (Ubl) domain interact with Ub. Most interactions involve five surface regions of the PLpro and two regions of Ub (Figure 2A). These five regions of the PLpro are labelled by Roman numbers: Ⅰ, Leu106-Tyr112; Ⅱ, Ala162-Arg168; Ⅲ, Cys208-Val210; Ⅳ, Gly248-Pro250; V, Phe269-Tyr279. Ⅰ and Ⅱ are situated in the thumb domain; Ⅲ is in the fingers domain; and Ⅳ and Ⅴ are in the palm domain (Figures 1A and 2A). In the following and in the figure labels, residues of Ub are indicated in italics to distinguish them from residues of PLpro. The two interacting regions of Ub are: A, Arg42-Gln49; B, Arg72-Gly76 (Figure 2A). Region A of Ub consists of β3, β4, and the loop between β3 and β4 (Figure 2B). Regions Ⅱ and Ⅲ of PLpro interact with region A of Ub. Region B comprises the five C-terminal residues, RLRGG, and is in contact with regions Ⅰ, Ⅱ, Ⅳ, and Ⅴ of PLpro(Figure 2C). The C-terminal RLRGG motif contributes the majority of the interactions with the PLpro; the buried surface between Ub region B and PLpro is 477 Å2(out of a total of 813 Å2). In order to make these interactions clear, we describe here in some detail the contacts between the PLpro and regions A and B of Ub.
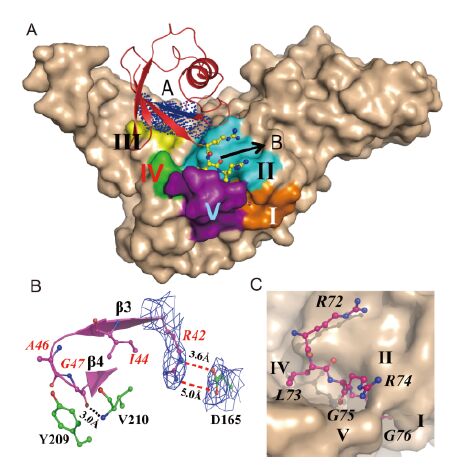
Figure 2. Interactions between MERS-CoV PLpro and Ub. (A) Five regions of PLpro bind to two areas of Ub. The surface of PLpro is shown in wheat color. The five interacting regions are shown in brown, cyan, yellow, green, and purple, and they are also labeled by Roman numbers. The two interacting areas of Ub are marked "A" and "B". Region A is highlighted by dark blue dots, and region B (the RLRGG sequence; Ub residues are in italics) is depicted in the ball-and-stick style. (B) Region A of Ub interacting with PLpro. A cartoon view of region A is shown in purple, and β3 and β4 are labeled. Residues of Ub (purple) and PLpro (green) are displayed in the ball-and-stick style, and labeled in red and black, respectively. The 2Fo-Fc electron density (blue; 1.0 σ) of the side-chains of Arg42 and Asp165 is displayed. Hydrogen bonds are indicated by black dashed lines, and the salt-bridge between R42 and D165 is depicted by two red dashed lines. (C) The RLRGG binding site of PLpro. The P5-P1 residues are shown in the ball-and-stick style. Regions Ⅰ, Ⅱ, Ⅳ, and Ⅴ that interact with the P5-P1 residues are labeled.
-
Region A (Arg42-Gln49) of Ub inserts into the space between the thumb and fingers domains of PLpro(Figures 2A-B). Residues Ile44, Ala46, and Gly47 engage in hydrophobic interactions with Tyr209 and Val210 of region Ⅲ of PLpro(Figure 2B). The hydrophobic patch (Ile44, Ala46, and Gly47) is a common interaction region utilized by Ub-binding proteins (Dikic et al., 2009). In particular, the interaction of Ile44 with Val210 of PLpro is important for the deubiquitinase (DUB) but not for the protease activity. The variant V210R shows dramatically reduced DUB activity, as demonstrated by Bailey-Elkin et al.(2014)(according to the numbering scheme of these authors, V210 is V1691).
A salt-bridge exists between region A of Ub and the PLpro(Figure 2B), namely between the side-chains of Arg42 and Asp165 (region Ⅱ in PLpro). In addition, a hydrogen bond is formed between the main-chain O atom of Gly47 and the main-chain amide of Val210 (region Ⅲ). In the SARS-CoV PLpro(C112S)-Ub complex (Chou et al., 2014), Arg42 forms a salt-bridge with the nega-tively charged Glu168, a residue which is replaced by the positively charged Arg168 in MERS-CoV PLpro. Consequently, the same salt-bridge cannot be formed in the MERS-CoV PLpro(C111S)-Ub complex. Instead, MERS-CoV PLpro has Asp165 interacting with Arg42. In our deubiquitinating (DUB) kinetic assays, the D165A variant shows a dramatically reduced DUB activity; the kcat/KM is about 78-fold decreased compared to that of the wild-type (Table 2). The KM value of the D165A variant is about 4-fold higher than that of the wild-type MERS-CoV PLpro, suggesting that this amino-acid replacement reduces the Ub binding affinity. Meanwhile, the D165E amino-acid replacement shows a catalytic efficiency comparable to wild-type towards Ub-7-amino-4-methylcoumarin (Ub-AMC) (Table 2). However, we noticed that the KM value of D165E for the DUB activity is about 2-fold larger than for the WT enzyme. Although Glu165 can mimic Asp165 here, we propose that the longer side-chain of Glu may fit less perfectly compared to Asp. These results indicate that the salt-bridge between Asp165 and Arg42 could be important for the PLpro's DUB activity, in addition to the interaction between Asp165 and the P4-amide group (see below).

Table 2. Kinetic parameters of MERS-CoV PLpro and SARS-CoV PLpro
-
Region B of Ub comprises the five C-terminal residues, RLRGG. These five residues bind to the narrow active-site channel between the thumb and palm domains of the PLpro(Figure 2C). They mainly interact with regions Ⅰ, Ⅱ, Ⅳ, and Ⅴ of the protease. Residues RLRGG are compati-ble with the PLpro cleavage motif, (R/K) (L/I) XGG (P5-P1), in the MERS-CoV polyproteins; their interactions with the PLpro are discussed here in terms of subsites S1 to S5.
-
Pro163 (region Ⅱ of PLpro) and the side-chains of Asn109 and Tyr112 (located in region Ⅰ) form a space-restricted S1 site to accommodate Gly76 (P1). The carbonyl oxygen atom of Gly277 (region Ⅴ) accepts a hydrogen bond from the amide of Gly76 (Figure 3).
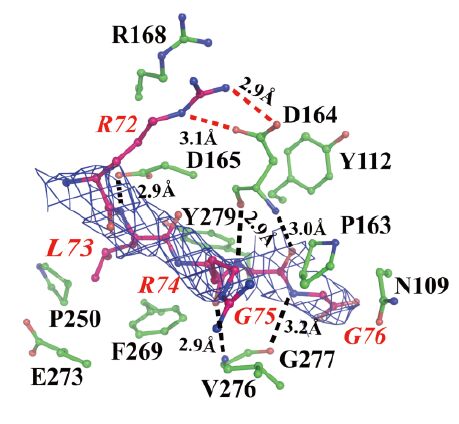
Figure 3. Details of the interactions between the C-terminal RLRGG of Ub and MERS-CoV PLpro. RLRGG residues are shown in purple in the ball-and-stick style, and they are labeled in red. For clarity, the 2Fo-Fc electron density (blue; 1.0 σ) of the RLRGG main chain is shown. PLpro residues are displayed in green in the ball-and-stick style, and they are labeled in black. Hydrogen bonds are displayed as black dashed lines, and the salt-bridge between R72 and D164 is depicted as two red dashed lines.
The side-chains of Tyr112 (region Ⅰ) and Phe269, Val276 as well as Tyr279 (region Ⅴ), and Gly277 form a restricted space for Gly75 (P2). Two hydrogen bonds link the main-chain at Asp164 of the PLpro and Gly75 (Figure 3). The S1 and S2 sites of the protease are too small to accommodate any other residue but glycine.
-
The main-chain O atom of Arg74 (P3) accepts a hydrogen bond from the Gly277 amide. In the complexes SARS-CoV PLpro(C112S)-Ub or SARS-CoV PLpro-Ubal, the main chain at Arg74 forms two hydrogen bonds (Chou et al., 2014; Ratia et al., 2014), namely with the amide of Gly272 and with the hydroxyl group of Tyr265. The former hydrogen bond is conserved in the MERS-CoV PLpro(C111S)-Ub complex, but the latter is not. Tyr265 of SARS-CoV is replaced by Phe269 in MERS-CoV, which lacks the ability to form a hydrogen bond with the main-chain amide of Arg74. In agreement with this difference, the DUB activity of SARS-CoV PLpro is 2.5-fold higher than that of MERS-CoV PLpro in our kinetic assay; furthermore, the MERS-CoV PLpro F269Y amino-acid replacement leads to enhancements by about 1.5-, 1.4-, and 1.9-fold of the hydrolytic activi-ties towards carbobenzoxy-Arg-Leu-Arg-Gly-Gly-7-amino-4-methylcoumarin (Z-RLRGG-AMC), Z-LRGG-AMC, and Ub-AMC, respectively (Table 2).
The side-chain of Arg74 is exposed to the solvent in our MERS-CoV PLpro(C111S)-Ub complex. This side-chain shows remarkable variability in the interactions it makes in the different complexes. In the "open" but not in the "closed" covalent MERS-CoV PLpro-Ub complex (Bailey-Elkin et al., 2014), it donates a hydrogen bond to the main-chain carbonyl oxygen of Thr1755 of the BL2 loop (corresponding to Thr274 in our numbering scheme). In SARS-CoV PLpro-Ubal (Ratia et al., 2014) but not in the SARS-CoV PLpro(C112S)-Ub complex (Chou et al., 2014), the side-chain of Arg74 forms a hydrogen bond with the main-chain carbonyl oxygen of Gln270. Instead, Arg74 is involved in a relatively weak salt-bridge with Glu162 in the SARS-CoV PLpro(C112S)-Ub complex (Chou et al., 2014). None of these interactions exist in our MERS-CoV PLpro-Ub complex.
-
The main-chain amide of Leu73 (P4) donates a hydrogen bond to the side-chain of Asp165 (region Ⅱ) (Figure 3). This hydrogen bond is conserved in all the complexes compared here. In our peptide-cleavage assay, the D165A variant shows about 16-fold and 10-fold lower activities towards substrates Z-RLRGG-AMC and Z-LRGG-AMC, respectively, indicating that Asp165 is not only important for interacting with Arg42 of Ub (see above).
The side-chain of Leu73 is embedded in a hydrophobic pocket which is formed by the Cβ atom of Asp165, the side-chain of Pro250 (region Ⅳ), Phe269, as well as the Cβ and Cγ atoms of Glu273 (region Ⅴ). Asp165 and Pro250 are conserved in SARS-CoV PLpro(Asp165 and Pro249). Phe269 is replaced by Tyr265, and Glu273, situated in the BL2 loop, is replaced by Tyr269. However, the side-chains of all these non-conserved residues possess the ability to provide a hydrophobic environment to accommodate Leu73.
-
The side-chain of Arg72 (P5) is located between the PLpro thumb domain and region A of ubiq-uitin. It forms a salt-bridge with the side-chain of Asp164 in MERS-CoV PLpro. In addition, the guanidinum group of Arg72 may be involved in a π-π interaction with that of Arg168 (Figure 3). These interactions have also been described for the covalent complex of MERS-CoV PLpro with Ub (Bailey-Elkin et al., 2014). Arg72 is not subject to strict space limitations; accordingly, this residue displays different binding patterns in the two SARS-CoV PLpro-Ub complexes. In the covalent SARS-CoV PLpro-Ubal complex (Ratia et al., 2014), Arg72 forms a salt-bridge with Glu168. In the non-covalent SARS-CoV PLpro(C112S)-Ub complex (Chou et al., 2014), Arg72 is exposed to the solvent and does not interact with Glu168(Arg42 instead forms a salt-bridge with Glu168, as mentioned above). In our kinetic assay, the D164A variant of MERS-CoV PLpro displays a ~4.5-fold and a ~3.5-fold reduced activity, respectively, for Z-RLRGG-AMC and Ub-AMC (Table 2). For Z-LRGG-AMC, the activity is decreased just a little (by about ~1.5-fold), because there is no P5-Arg in this substrate (Table 2). These data demonstrate that Asp164 is important for the interaction with Arg72.
In summary, the main-chain heteroatoms of P5-P1 form a hydrogen-bonding network with PLpro. The binding characteristics of P1, P2, and P4 are conserved in all MERS-CoV and SARS-CoV PLpro-Ub complexes. However, P3-Arg and P5-Arg assume binding patterns that differ between the various MERS-CoV and SARS-CoV PLpro-Ub complexes.
Overall structure of MERS-CoV PLpro in complex with human ubiquitin
Interactions of MERS-CoV PLpro(C111S) with Ub region A
Interactions of MERS-CoV PLpro(C111S) with Ub region B
S1 and S2 subsites
S3 subsite
S4 subsite
S5 subsite
-
Viral proteins are likely to possess multiple functions, as exemplified by non-structural protein 1(NS1) of influenza A viruses (Hale et al., 2008), the nucleocapsid (N) protein of coronaviruses (Chang et al., 2014), or the Nsp14 exonuclease-guanyl-7-methyltransferase of coronaviruses (Chen et al., 2009). Exhibiting DUB and proteolytic activities, the papain-like proteases of coronaviruses are no exception here. Although the overall folds are conserved, the enzyme activity and substrate-binding modes of CoV PLpros differ in detail. Therefore, no coronavirus PLpro can be considered a general model for all its homologues (Baez-Santos et al., 2014b).
-
We have previously noticed that the oxyanion hole of the MERS-CoV PLpro appears to be deficient (Lei et al., 2014; also see the discussion below). Similarly, the recognition of ubiquitin by the enzyme appears to be sub-optimal. Thus, the main-chain amide of the P3-Arg residue has no hydrogen-bonding partner on the MERS-CoV PLpro, because the near-by side-chain of Phe269 is incapable of accepting an H-bond. The corresponding residue is Tyr265 in the SARS-CoV PLpro, which is perfectly positioned to accept the hydrogen bond from the P3-amide. Indeed, when we replaced Phe269 by Tyr in MERS-CoV PLpro, the DUB activity of the enzyme increased by a factor of almost 2 and the peptidolytic activity by ~1.5. The evolution of viral enzymes is not necessarily driven by optimization of catalytic efficiency. This is particularly true for viral proteases that have to ensure the availability of non-structural proteins in the correct temporal order when they cleave them out of the viral polyproteins; in fact, too rapid a polyprotein processing might be counterproductive. On the other hand, a more efficient DUB activity should help the virus in counteracting the innate immune response of the host cell. As the binding of the P5-P1 residues of ubiquitin and of the polyprotein cleavage sites obviously influences both the DUB and proteolytic activities of the PLpro, the sub-optimal catalytic efficiency that we observe may be a consequence of a compromise between the requirements of the two activities.
With regard to the oxyanion hole, we previously proposed that the backbone amide of Asn109(located in a β-turn connecting β7 and α4, Figure 4) may contribute to the stabilization of the oxyanion intermediate in PLpro catalysis, along with the main-chain amide of the active site Cys111, although this may require a slight rearrangement of this β-turn (Lei et al., 2014). In our non-covalent MERS-CoV PLpro-Ub complex, we do not see any rearrangement of the β-turn. More or less in agreement with our suggestion, Bailey-Elkin et al.(2014)proposed on the basis of their covalent MERS-CoV PLpro-Ub complex structure that the main-chain amides of Asn1590, Asn1591, and Cys1592(corresponding to Asn109, Asn110, and Cys111 in our numbering scheme) form the oxyanion hole. On the other hand, Lee et al.(2015)argued that the side-chain of Asn109 could contribute to the oxyanion hole. They found that the N109A replacement completely abolished enzyme activity. As we reported earlier (Lei et al., 2014), the side-chain amide of Asn109 makes a strong hydrogen bond with the conserved Gly161(Figure 4). Any reorientation of the Asn109 side-chain towards the oxyanion would require a disruption of this strong interaction; this is not very likely. In conclusion, lacking a side-chain in the proper spatial orientation and capable of donating a hydrogen bond to the oxyanion (such as Trp107 in SARS-CoV PLpro), the oxyanion hole of MERS-CoV PLpro seems to be deficient.
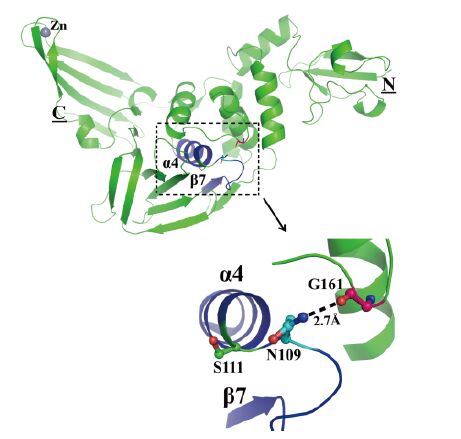
Figure 4. The side-chain of Asn109 is unlikely to contribute to the oxyanion hole of MERS-CoV PLpro. PLpro is shown in green in cartoon view. N and C termini are marked by underlined letters. Strand β7, helix α4, and the loop between them are colored in blue. Asn109 and Gly161 are indicated in ball-and-stick style in cyan and purple, respectively, and they are labeled in black. The hydrogen bond between them is displayed as a black dashed line. The active-site Cys111 (replaced by Ser in this study) is shown in green in ball-and-stick style.
-
Apart from the less than optimum binding of the P3 residue, there are other differences in the way MERS-CoV PLpro and SARS-CoV PLpro recognize human ubiq-uitin. The formation of a salt-bridge between Arg72, the P5 side-chain of Ub, and Asp164 is unique for MERS-CoV PLpro. The same interaction exists in the two covalent MERS-CoV PLpro-Ub complexes (Bailey-Elkin et al., 2014). This binding mode is very different from the Glu168 -Arg72 salt-bridge in the SARS-CoV PLpro-Ubal complex (Ratia et al., 2014). Glu168 is conserved in HCoV-NL63 PL2pro and replaced by Asp in the HCoV-229E, HCoV-OC43, and HCoV-HKU1 PL2pros (for sequence alignments, see Barretto et al., 2005; Baez-Santos et al., 2014b), so the same type of interaction is likely to be realized in the Ub complexes of these enzymes. However, the corresponding residue in MERS-CoV PLpro is Arg168; hence, Asp164 is used instead to bind Arg72. Asp164 is in fact unique in MERS-CoV. It is replaced by Gly in SARS-CoV PLpro and the PL2pros of HCoV NL63 and HCoV 229E, by Ala in HCoV-OC43 PL2pro, and by Ser in HCoV-HKU1 PL2pro. Our kinetic results for the D164A replacement (see Results) emphasize the impor-tance of the unique Asp164 residue.
-
In our PLpro(C111S)-Ub complex, the Ubl domain of PLpro shows no interaction with ubiquitin. The relative orientation of the Ubl domain in the substrate-bound PLpro is the same as in substrate-free PLpro. The Ubl domain of MERS-CoV PLpro is not required for the IFN antagonism activities (Baez-Santos et al., 2014b), but it is required in case of SARS-CoV PLpro(Frieman et al., 2009). Recently, Mielech et al.(2015)reported that the Ubl domain of mouse hepatitis virus (MHV) is an important modulator of PLpro stability and viral pathogene-sis. Although the Ubl domain shows variable effects in different CoVs, the high degree of conservation of the domain throughout the family suggests that it may play a common biological role. One possible function is that the Ubl might be involved in protein-protein interactions. Ubiquitin-like domains are known to function as binding modules in such interactions. For example, the kinase Raf contains a Ubl domain for interaction with human Ras (Fetics et al., 2015). Also, human ubiquitin-specific protease (USP)7 includes five Ubl (1-5) domains, of which the second is bound by the Herpes simplex virus-1 immediate-early protein ICP0 to counteract the intrinsic antiviral response of the host cell (Pfoh et al., 2015).
-
The BL2 loop of SARS-CoV PLpro is important for binding inhibitors (Baez-Santos et al., 2014a; Lee et al., 2015). This loop is variable in different CoV PLpros. A potent inhibitor of the SARS-CoV PLpro, N-[(3-fluo-rophenyl) methyl]-1-[(1R)-1-naphthalen-1-ylethyl] piperi-dine-4-carboxamide (compound 3k, IC50 = 0.15 ± 0.01 μmol/L) was found to have no effect on the MERS-CoV enzyme (Baez-Santos et al., 2014b). Tyr269 and Gln270 of SARS-CoV PLpro, which are important for binding this inhibitor (Baez-Santos et al., 2014a), are replaced by Glu273 and Ala275 in the MERS-CoV protease. It seems that this structural difference in the BL2 loop has a remarkable impact on the effectiveness of the inhibitor. The structure of the MERS-CoV PLpro(C111S)-Ub complex presented here will facilitate virtual screening of chemical libraries for specific anti -MERS-CoV PLpro inhibitors (Hilgenfeld, 2014).
-
SARS-CoV and MERS-CoV PLpros can digest K48-and K63-linked (poly) ubiquitin chains and remove ISG15 from proteins covalently linked to it (Ratia et al., 2014, Baez-Santos et al., 2014b). SARS-CoV PLpro prefers binding of K48-Ub2 and ISG15 over mono-ubiquitin (Ratia et al., 2014). All evidence suggests that on the PLpro, at least two ubiquitin-binding sites (or a binding site for diubiquitin-like molecules such as ISG15) exist. Ratia et al.(2014)proposed hypothetic models for complexes of SARS-CoV PLpro with K48-Ub2 and ISG15. These authors identified two major hydrophobic binding sites on the PLpro for the first Ub (Ub1) and the second Ub (Ub2). The binding site for Ub1 comprises Met209, Pro248, and Pro249. This hydrophobic patch is conserved in MERS-CoV PLpro, although the residues are not exactly the same. Met209 of SARS-CoV PLpro is replaced by Val210(region Ⅲ) in MERS-CoV. Pro248 is replaced by Thr249, whereas Pro249 is conserved (Pro250; region Ⅴ) (Figure 5). The hypothetic second binding site for Ub2(also named "ridge" region, Ratia et al., 2014) is located to the first α helix (α2) in the thumb domain, including residues Phe70, His74, and Leu76 of SARS-CoV PLpro. According to a structural alignment of the SARS-CoV and MERS-CoV PLpros, Phe70, His74, and Leu76 are changed to Lys69, Gly73, and Val75, respectively (Figure 5). In SARS-CoV PLpro, the F70S and F70A replacements lost the affinity to K48-Ub2 and ISG15 in vitro (Ratia et al., 2014); therefore, the presence of Lys69 in MERS-CoV PLpro instead of Phe70 strongly suggests that this enzyme should bind Ub2 in a different way. Bailey-Elkin et al.(2014)predicted Asn1673 and Val1674 of the fingers subdomain (corre-sponding to Asn192 and Val193 in our PLpro) as the distal Ub site of K63 di-Ub, but they found that their DUB activity data do not support their prediction for the Ub2 binding site. The structure of the MERS-CoV PLpro-Ub complex reported here reveals the exact Ub1 binding site on the PLpro, but the Ub2 binding site should be identified by crystallizing the PLpro in complex with poly-or di-ubiquitin.

Figure 5. The two Ub-binding sites of PLpro. The Ub1 and the proposed Ub2 (according to a structural alignment with the SARS-CoV PLpro; Ratia et al., 2014) binding sites are depicted as purple dots. The N and C termini of PLpro are marked by underlined letters. All residues related to the two Ub binding sites are labeled. The catalytic triad Cys111-His278-Asp293 is indicated by yellow, blue, and red spheres.
In summary, the crystal structure of the MERS-CoV PLpro-Ub complex provides valuable information that helps understand the multiple functions of coronavirus papain-like proteases. Mutational studies additionally highlight features of the MERS-CoV PLpro. The different substrate-binding patterns should be kept in mind when designing inhibitors for PLpros of CoVs, even though the overall structures of these enzymes are conserved. Furthermore, it would be helpful to obtain the structure of PLpro in complex with di-Ub or ISG15 in the future.
MERS-CoV PLpro is not optimized for catalytic efficiency
Unique features of Ub recognition by MERS-CoV PLpro
The role (s) of the ubiquitin-like (Ubl) domain
PLpro inhibitors
The di-Ub site of PLpro
-
Technical assistance by Susanne Zoske is gratefully acknowledged. We thank Stefan Anemüller for discussion. We also acknowledge access to beamline BL14.2 operated by the Helmholtz-Zentrum Berlin at the BESSY Ⅱ electron storage ring (Berlin-Adlershof, Germany). This work was supported by the European Commission through its "SILVER" project (contract no. HEALTH-F3-2010-260644) and by the German Center for Infection Research (DZIF). RH acknowledges support by the DFG Cluster of Excellence "Inflammation at Interfaces"(EXC 306)
-
This article does not contain any studies with human or animal subjects. Both authors declare no competing interest.
-
JL and RH designed all experiments. JL performed the experiments. JL and RH analyzed all data. JL and RH wrote the manuscript.
Supplementary figure and table are available on the websites of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-

Figure S1. Kinetics of Ub-AMC hydrolysis by MERS-CoV PLpro (WT) and its variants. Wild-type: WT; reaction rate: V (unit: μmol/L/min); enzyme concentration: [E] (unit: μmol/L).
Primers C111S 5′-CAAATTGAGTGATAATAATTCTTATCTTAATGC 3′-CATAATAACTGCATTAAGATAAGAATTATTATCACTC F269Y 5′-GTAGCATTTAATGTCTATCAGGGCATTGAAACG 3′-CGTTTCAATGCCCTGATAGACATTAAATGCTAC D165E 5′-GGTGCTCCAGATGAGGCCTCTCGGTTACTT 3′-AAGTAACCGAGAGGCCTCATCTGGAGCACC D165A 5′-GGTGCTCCAGATGCTGCCTCTCGGTTACTT 3′-AAGTAACCGAGAGGCAGCATCTGGAGCACC D164E 5′-ACATTTGGTGCTCCAGAGGATGCCTCTCGGTTA 3′-TAACCGAGAGGCATCCTCTGGAGCACCAAATGT D164A 5′-ACATTTGGTGCTCCAGCTGATGCCTCTCGGTTA 3′-TAACCGAGAGGCATCAGCTGGAGCACCAAATGT Table S1. ll primers of MERS-CoV PLpro mutants

 DownLoad:
DownLoad: