










-
Restriction endonuclease analysis (REA), or restriction fragment length polymorphism (RFLP), was useful for identifying and determining the relatedness and putative identities of microbial strains (Tang et al., 1997) and for characterizing and discriminating large numbers of samples inexpensively in the past, including human and simian adenoviruses (Li et al., 1986). However, the low-resolution data limited its applications, as REA assays can be subjective in terms of performance and interpretation; its value is dependent on the user’s judgment as well as experimental conditions. Specifically, the empirical choices of reference genomes and restriction enzymes affect the interpretation, and may be further muddled by confirmation bias. Experimentally, contaminating DNA and incomplete restriction enzyme digestions also play roles in misinterpretations, as illustrated by the misidentification and typing of HAdV-B14p1 as HAdV-B14a initially (Houng et al., 2010; Kajon et al., 2010; Louie et al., 2008; Metzgar et al., 2007). REA is not used widely now and, anecdotally, this technique is considered obsolete in the current era of relatively low-cost, cost-efficient, and high-resolution genome sequencing. However, it is still a potentially useful, rapid, and inexpensive method that is still appropriate for screening large numbers of samples, once the pitfalls are recognized. It is also invaluable and essential for characterizing current isolates by providing a bridge between their accessible genome data and the REA data that are only available in the literature for important reference and historical isolates that are no longer available for genomic analysis. This is important for understanding the molecular epidemiology and evolution of viral pathogens, particularly with periodically re-emergent strains.
A recent controversy over the naming of a human adenovirus (HAdV) type 55 as type "11a" (Kajon et al., 2013; Walsh et al., 2010) serves as instructive example to illustrate some of the pitfalls of relying on REA to identify, characterize, type, and name adenoviral pathogens in the era of whole genome data. Some of these concerns have been addressed earlier specifically for adenovirus characterization, where it was noted REA is useful for "prototype-like restriction patterns" but "the occurrence of genome types with deviating restriction patterns limits the application of this method" (Wigand, 1987). One important example of these "deviating restriction patterns" is a recombinant genome. Another example of the ambiguities of REA data interpretation is presented in mis-characterizations of HAdVs due to sample and electrophoresis gel quality, and the resultant interpretation of REAs. To provide additional support for using REA data correctly and to highlight some of the caveats of interpretation and application within the context of identifying, characterizing, typing, and naming HAdVs with REA, this report presents the computational analyses, including in silico REA, of two contemporaneous circulating genomes of HAdV-B55 in the context of the proper reference genomes of HAdV-B11 and B14. Thus, given these caveats and corrections, REA is still applicable and useful in this era of genomics.
The genomes of HAdV-B11p, HAdV-B14p, HAdV-B14p1, and HAdV-B55 are accessible from GenBank (accession number): HAdV-B11 (AY163756), HAdV-B55 QS-DLL (FJ643676), HAdV-B55 SGN1222 (FJ597732), HAdV-B14p (AY803294), and HAdV-B14p1 (FJ822614). These genomes span approximately sixty years. Table 1 presents the genome percent identities of these HAdVs, highlighting a difficulty in discriminating these highly similar viruses from each other.
HAdV-B55 SGN1222 HAdV-B11p HAdV-B14p HAdV-B14p1 HAdV-B55 QS-DLL 99.8 97.6 98.8 98.8 HAdV-B55 SGN1222 97.6 98.9 98.9 HAdV-B11p 97.2 97.2 HAdV-B14p 99.7 Note: Several subspecies B2 human adenoviruses are nearly identical to each other with regards to their genome sequences, as noted by genome percent identities. Small recombinant regions do not affect the overall percent identity to a significant degree. This presents difficulties in discriminating each individual virus. These values were calculated using the software EMBOSS Stretcher (http://www.ebi.ac.uk/Tools/psa/emboss_stretcher/nucleotide.html). Table 1. Genome percent identities
Despite the overall high degree of genome identities, high-resolution inspection of the DNA sequences allows discrimination of each unique virus using genomics. As an example, a portion of the phylogenetic tree comprising all HAdV genomes is presented in Figure 1. Within the clade of HAdV subspecies B2, HAdV-B11p branches away from both HAdV-B14 and B55, which is represented by the QS-DLL strain; these latter two HAdVs form a subclade. These branches are significant as the bootstrap values are above 80. This subclade reflects the published comparative genomics and recombination detection results showing that HAdV-B55 arose from a recombination event between HAdV-B11 and HAdV-B14, resulting in a novel respiratory pathogen (Walsh et al., 2010).
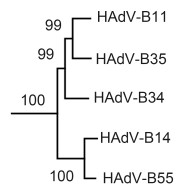
Figure 1. Phylogenetic clade of HAdV subspecies B2. Whole genome data from all HAdVs, available from GenBank were downloaded, aligned, and subjected to phylogenetic analysis. MEGA4 was used to construct bootstrapped, neighbor-joining trees with 1, 000 replicates (http://www.megasoftware.net/mega4/mega.html). A portion of the larger phylogenetic tree is presented to highlight the members of HAdV subspecies B2. This, and phylogenetic trees of individual genes (data not shown), demonstrate that these viruses may be discriminated individually based on the genome sequence despite near identities in genome identity percentages. All genomes are from the representative prototype viruses: HAdV-B55 is the QS-DLL index strain. Bootstrap values above 80 are considered robust.
To mirror and re-evaluate a report which was reliant, in part, upon wet-bench experimental REAs for the identification, characterization, and typing of several HAdV subspecies B2 viruses as variants of HAdV-B11p (Kajon et al., 2013), the following restriction enzymes (REs) were selected for in silico evaluation: BamH Ⅰ, Bcl Ⅰ, Bgl Ⅱ, BstE Ⅱ, Hind Ⅲ, Hpa Ⅰ, Pst Ⅰ, Sma Ⅰ, and Xba Ⅰ. One caveat, and a key contrast to the gel data published (Kajon et al., 2013), is that appropriate and correct reference genomes are provided in this in silico re-analysis, that is, HAdV-B11p and HAdV-B14p. This significant requirement for appropriate REA reference genomes was also noted in a review of techniques for characterizing HAdVs by Wigand (Wigand, 1987).
BamH Ⅰ REA shows HAdV-B14p and B14p1 as having identical band patterns. Importantly, these are also identical to the REA profiles for HAdV-B55, QS-DLL and SGN1222 (Figure 2A, Panel A). In contrast, these differ from the HAdV-B11p BamH Ⅰ RE map, which was taken as the basis for identifying, characterizing, and naming HAdV-B55 as a genome type and variant of HAdV-11 ("HAdV-11a") (Kajon et al., 2013). Panel B presents the Bcl Ⅰ REA patterns in which HAdV-B14p1 is nearly identical to both HAdV-B55 REA patterns with one band size exception, which may not be obvious on a wet-bench gel (band 2 of lanes b and c is 5962 bp; and band 2 of lanes d and e is 5958 bp). This is one important caveat to using REAs: while in silico data have a quantitative discrimination of single base changes, the wet-bench version is highly dependent on the user’s eyesight, gel electrophoresis conditions, and subjective-perhaps biased-view. Larger indels may also present misleading patterns and interpretations if the assumption is that REA differences are due solely to changes in the RE sites. Doublets are represented as "fuzzy" thicker bands, which may be missed also by visual inspection of wet-bench gels. For the Bcl Ⅰ RE assays, HAdV-B14p1 shares bands with HAdV-B55, while displaying three banding differences from HAdV-B14p (noted by the arrows). Bcl Ⅰ allows the two HAdV-B14 genomes to be differentiated from each other, as does BstE Ⅱ. Panel C shows Bgl Ⅰ band patterns in which both HAdV-B14p and B14p1 have identical patterns to the profiles for HAdV-B55 QS-DLL and SGN1222. These patterns differ from the HAdV-B11p Bgl Ⅰ RE map. Panel D presents the BstE Ⅱ RE patterns in which HAdV-B14p and B14p1 may be discriminated from each other using this enzyme. The HAdV-B55 patterns are identical to each other and similar to B11p, but are subtly different, with four band shifts (numbers 1, 2, 6, and 7) and one missing band.

Figure 2. In silico REA in five closely related HAdV genomes. HAdV-B11 (a); HAdV-B55 QS-DLL (b); HAdV-B55 SGN1222 (c); HAdV-B14p (d); and HAdV-B14p1 (e) are assayed using nine restriction enzymes and Vector NTI Advance 11.5 (Invitrogen Corp.; San Diego, CA. USA). (A): BamH Ⅰ (Panel A), Bcl Ⅰ (Panel B), Bgl Ⅱ (Panel C), and BstE Ⅱ (Panel D); (B): Hind Ⅲ (Panel A), Hpa Ⅰ (Panel B), and Pst Ⅰ (Panel C); (C): Sma Ⅰ (Panel A) and Xba Ⅰ (Panel B). Arrows highlight differences between HAdV-B14p, HAdV-B14p1 and B55 patterns. Size markers are noted in "M".
Hind Ⅲ, Hpa Ⅰ, and Pst Ⅰ REAs are presented in Figure 2B, with Panel A (Hind Ⅲ) providing identical patterns for HAdV-B14p and B14p1; HAdV-B55 strain QS-DLL shows a band difference with strain SGN1222, which may not be discriminated on a wet-bench gel, and both are missing the three bands contained in the HAdV-B14 RE band patterns. HAdV-B11p shows similarity with both HAdV-B55 and B14 patterns using Hind Ⅲ. In Panel B (Hpa Ⅰ), HAdV-B14 and B14p1 are identical and are similar to both HAdV-B55 patterns. In contrast, the HAdV-B11p RE pattern appears to be very different, albeit with six possible common bands with the other RE patterns. In Panel C (Pst Ⅰ), HAdV-B14p and B14p1 provide similar yet different patterns: At four band positions, HAdV-B14p is more similar than HAdV-B14p1 to the HAdV-B55 patterns (noted by the arrows). Pst Ⅰ allows for the discrimination of the two nearly identical HAdV-B14 genomes.
Sma Ⅰ and Xba Ⅰ REA patterns, shown in Figure 2C, highlight how similar all of these genomes are, with Panel A (SmaⅠ) displaying nearly identical patterns for all of the HAdV-B14 and B55 genomes. HAdV-B11p shares perhaps five bands, but differs at seven bands. Panel B highlights differences between HAdV-B55 QS-DLL and SGN1222, with three band differences (noted by the arrows); SGN1222 is identical to the patterns for both HAdV-B14 genomes, whereas QS-DLL is different. In this case, a point may be made that QS-DLL should be renamed HAdV-B55p1, analogous to HAdV14p1 and according to Li and Wadell’s genome type denomination system (Li et al., 1986). Nota bene, the rules regarding genome type denomination are not clearly stated, and are qualitative and arbitrary, as illustrated by the REA patterns presented in this report and in earlier reports as well (Kajon et al., 2010; Kajon et al., 2013; Li et al., 1986).
REA was useful in the past, e.g., screening large numbers of samples rapidly. It still is a useful, rapid, and relatively inexpensive technique, particularly in laboratories that are not equipped for genome sequencing and other costlier techniques. However, these seemingly simple and clear data must be evaluated with caution, as noted earlier by Wigand (Wigand, 1987) and demonstrated in this report. Certain caveats need to be taken into account as overreaching and incorrect conclusions may be drawn from the interpretation of the data. One example of the ambiguities of REA gel data interpretation, ironically, is a report by Curtis et al., in which earlier reported serotyping of several epidemic strains was described as "unsatisfactory, wrongly assigning the isolate to serotype 10" by the investigators (Curtis et al., 1998). The investigators presented REA data as evidence that the strain was actually serotype 37; however, the experimental data presented were not convincing and were difficult to interpret, with the RE banding patterns of varying staining intensities and sizes (Curtis S et al., 1998).
Recent commentaries point to the value and promise of whole genome data and analysis in providing high-resolution insights of microbes in public health (Relman, 2011), including clinical virology (Cruz-Rivera et al., 2013). The emergent or re-emergent ARD pathogen HAdV QS-DLL was isolated and its genome sequenced (Yang et al., 2009; Zhu et al., 2009) and, when re-analyzed using computational methods, including phylogenetics (Figure 1) and recombination detection software (data not shown) (Walsh et al., 2010), QS-DLL was found to contain a unique genome with only the epsilon epitope derived from HAdV-B11, a renal pathogen, comprising 2.6% of the genome and contributing to virus neutralization in serological assays, embedded in the genome chassis of HAdV-B14, a respiratory and ARD pathogen. As a result, this novel virus was renamed HAdV-B55 (Walsh et al., 2010) in consultation with the authors of the original reports (Yang et al., 2009; Zhu et al., 2009) and in accordance with a proposal accepted by a majority of the adenovirus research community to use the whole genome data rather than serological data or only the epsilon epitope data as a means to identify, name, and type HAdVs (Seto et al., 2011). This was reaffirmed in discussions at the 10th International Adenovirus Meeting (Umea, SWE; 2012), with the consensus being that "genotype" refers to adenoviruses described and type-numbered with genome data and "molecular type" is used to name viruses with only limited DNA sequence data. "Serotype" is reserved for HAdVs characterized, as in the past, by thorough and complete bench-run serological assays, including both VN and HI assays, and heterologous titrations with antisera generated against the query HAdV. Therefore, the name for genotype "HAdV-B55" is correct, identifying a novel fully sequenced virus with a unique genome and representing a new prototype. Presented in this report are REA results supporting this identification as well.
That HAdV-B55 is a novel and unique virus and human pathogen is illustrated by its biology and clinical attributes. It is a "Trojan Horse" pathogen as its immunological epitope (epsilon) corresponds to a renal pathogen, but its tropism is to the lungs and it is a respiratory pathogen (Walsh et al., 2010). Its parental genomes are HAdV-B11p (Kibrick et al., 1957) and HAdV-B14p (Van Der Veen et al., 1957) which were isolated in the 1950s, and are renal tract and respiratory tract pathogens, respectively. There are other examples of other recently isolated emergent and "Trojan Horse" viral pathogens characterized by whole genome analysis and containing recombinant genomes include HAdVs with an immunological epitope (nonpathogenic) that contrasts with their pathogenic and phenotypic properties (epidemic keratoconjunctivitis). Two examples include HAdV-D53 (Walsh M P et al., 2009) and HAdV-D64 (Zhou et al., 2012). In contrast, these genotypes and their names have not generated the same controversy as HAdV-B55, although the recombination events, e.g., a lateral transfer of a partial gene sequence comprising 2.6% of the whole genome, are very similar and demonstrate genome recombination is an important molecular evolution mechanism by which novel HAdV pathogens emerge.
As observed in the in silico REA/RFLP analysis of HAdV-B55, caveats should be carefully considered to ensure that the data and results are meaningful and interpreted correctly. For clarity, again, characterization and proposed naming HAdV-B55 as type "11a" is incorrect due to the confirmation bias and incorrect application of the REA method; it is a novel and unique HAdV that is correctly named HAdV-B55 (Kajon et al., 2013; Walsh et al., 2010). It is unfortunate the so-called "11a" virus is no longer available and that there are no published REA data for comparisons. First and foremost of the caveats is the recognition and application of appropriate "controls", i.e., reference genomes. If a query HAdV is to be considered as a variant of a prototype, then that prototype must be included for comparison and the REA patterns should reflect the proposed relationship. In this report, presented in Figure 2 are the in silico REA data for two HAdV-B55 genomes along with the correct and necessary reference genomes, i.e., parental HAdV-B11p and HAdV-B14p genomes. The in silico REA patterns of HAdV-B55 SGN1222 are identical to those of the proposed and incorrectly named "HAdV-11a" (Kajon et al., 2010; Kajon et al., 2013); when HAdV-B14p is included as a reference appropriately, it is clear that both HAdV-B55 genomes show much less REA pattern similarities to HAdV-B11p and near identities to HAdV-B14p, as would be expected given the whole genome data. Again, for HAdV-B55 to be considered a genome type variant of HAdV-B11p, i.e., "HAdV-B11a", it must have a genome that is based upon and similar to HAdV-11p, similar to the survey and definition of HAdV-B7 genome type variants by Li and Wadell (Li et al., 1986). Ironically, given the near identities of the HAdV-B55 REA patterns to their counterparts in HAdV-B14 for six enzymes (BamH Ⅰ, Bcl Ⅰ, Bgl Ⅱ, Pst Ⅰ, Sma Ⅰ, and Xba Ⅰ), an argument could be made for naming HAdV-B55 a genome type variant of HAdV-B14. This detracts from the reality that it is, again, a unique and novel, emergent human viral pathogen. The REA patterns for the other three enzymes provide arguments for HAdV-B55 being similar to either HAdV-B14 or HAdV-B11 (BstE Ⅱ, Hind Ⅲ, and Hpa Ⅰ), and demonstrate the confirmation bias "supporting" the ambiguous partial serological typing data (Kajon et al., 2013).
Along these lines, another caveat is that REA is not useful nor is appropriate for analyzing and characterizing recombinants. It is difficult to determine the appropriate reference and prototype genomes, without whole genome analysis, as demonstrated for HAdV-B55 and as presciently cautioned by Wigand nearly two and a half decades ago (Wigand, 1987).
To add further support to this, another example of these pitfalls of relying on REA data is illustrated by another recent misidentification/mistyping of a re-emergent HAdV respiratory pathogen based on REA as well: the characterization of the ARD pathogen HAdV-B14p1 (Metzgar et al., 2007). This pathogen reemerged in 2007 after an absence of ca. 50 years (Metzgar et al., 2007; Van Der Veen et al., 1957). REA patterns, using BamH Ⅰ, Bgl Ⅱ, Hind Ⅲ, and Sma Ⅰ, established this pathogen as a "new genome type that we have designated Ad14a" (Louie et al., 2008) with subsequent publications (Wang et al., 2009). However, the wet-bench REA analysis was flawed, with the original additional bands attributed subsequently to contaminating DNA, and the strain has been subtly renamed HAdV-B14p1 (Kajon et al., 2010) after final whole genome sequence determination (Houng et al., 2010). Additionally, REA patterns are not always correct/informative as the comparative genomic analysis of four HAdV-B14 strains indicated (manuscript submitted).
One additional pitfall is that REA is low resolution, that is, it samples only selected RE sites. In the context of comparing 35 kb genomes, these sites, which may be few and far apart, are not entirely adequate. In contrast, a comprehensive mutations analysis, given a compete genome sequence, may serve to ascribe lineages more accurately and in greater detail, particularly marking iconic insertion and deletion (indels) events.
In conclusion, the REA method is a technique that was useful in the pre-genomics era and is still useful today for inexpensively and rapidly screening large numbers of samples, particularly from an outbreak or several related outbreaks. Potentially interesting samples found by this initial screen may be further genome-sequenced for additional insights. In this scenario, REA should be used along with the partial DNA sequencing of the relevant genetic markers to ensure correct applications of reference genomes. Obviously, genomic sequencing provides more information than REA, and REA can only identify one or several nucleotide mutations within RE sites. Whole genome sequencing will be tenable once costs are lowered and more accurate genome assembly software is available. For now, in the context of appropriate references including genome sequences, a rapid and relatively inexpensive survey of many isolates is possible using REA, a still useful technique for characterizing viral pathogens.
HTML
-
This work was supported by the National Natural Science Foundation of China (31570155 and 31370199) and "Young Top-notch Talents" of the Guangdong Province Special Support Program (2014), as well as the Excellent Young Teacher Training Plan of Guangdong Province (Yq2013039) and the Guangzhou Healthcare Collaborative Innovation Major Project (201400000002). Portions of this manuscript were completed at the Department of Ophthalmology, Howe Laboratory, Massachusetts Eye and Ear Infirmary, Harvard Medical School (Boston, Massachusetts, USA) as Q.Z. was funded by the China Scholarship Council (CSC No. 201508440056) as a Visiting Scholar (2015-2016); he thanks Professor James Chodosh for providing a stimulating intellectual environment. The project was additionally supported by a summer research grant to D.S. from the Office of the Vice President for Research at George Mason University. The authors declare that they have no conflict of interests. This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: