









-
Rodents are a harbor of several zoonotic pathogens that may be transmitted to humans via direct or indirect contact. Some of these viruses are closely related to human viruses, and are therefore suspected to possess zoonotic potential (Luis et al., 2013). Among these viruses, zoonotic transmission of mammarenavirus and hantaviruses has already been demonstrated.
Mammarenaviruses (genus Mammarenavirus, family Arenaviridae) are serious pathogens associated with hemorrhagic fever and diseases of the central nervous system. Rodents are the primary natural reservoir of mammarenaviruses (Gonzalez et al., 2007; Charrel and de Lamballerie, 2010; Radoshitzky et al., 2015). In 2014, the Wenzhou virus (WENV), which is carried by rodents and shrews in Wenzhou city, Zhejiang province, China, was isolated and characterized (Li et al., 2015). In 2016, the Cardamones variant of WENV, which was found in Cambodia and Thailand, was shown to be associated with human infection (Blasdell et al., 2016).
Hantaviruses (genus Orthohantavirus, family Hantaviridae, order Bunyavirales) have been shown to cause two main diseases in humans: hemorrhagic fever with renal syndrome (HFRS) in Eurasia, and hantavirus cardiopulmonary syndrome in North America (Adams et al., 2017). Rodents are the natural hosts of hantaviruses; humans are accidental hosts and are infected by aerosols of infected rodents (McCaughey and Hart, 2000). HFRS is a severe public health issue in China; incidences of HFRS in this country have accounted for the highest proportion (approximately 90%) of cases globally in the last few decades (Huang et al., 2012). In China, two specific hantaviruses cause HFRS: Hantaan virus (HTNV), which is harbored by the striped field mouse (Aodemus agrarius), and Seoul virus (SEOV) harbored by the Norway rat (Rattus norvegicus) (Jiang et al., 2017).
Hepatitis E virus (HEV) is the prototype of the family Hepeviridae. HEV infections are self-limiting, and the clinical course of symptomatic HEV infections includes acute and chronic hepatitis E, fulminant liver failure, and extrahepatic symptoms (Kamar et al., 2012; Smith et al., 2014). Domestic pigs and wild boars have been recognized as the main animal reservoirs of zoonotic HEV, although deer and camel HEV may also be transmitted to humans (Meng, 2010; Lee et al., 2016). Currently, molecular detection methods have revealed HEV-related isolates in wild rats, shrews, and bandicoots from Zhanjiang city, Guangdong province, China, which might indicate a broader host range or spillover infections (Guan et al., 2013; Li et al., 2013). However, the zoonotic potential of rodent and shrew HEV remains controversial (Johne et al., 2014).
As humans are often in close contact with rodents and shrews, viruses harbored by these animals represent a threat to human health. Shenzhen is a major city in southern China, with an area of 40,000 km2 and a population of 11 million. The city is located directly north of the Hong Kong special administrative region and holds sub-provincial administrative status. Shenzhen city has been reported to be an important endemic area for HFRS caused by SEOV harbored by Norway rats (Yang et al., 2006; Liu et al., 2008). However, little is known about the presence of mammarenavirus and HEV in rodents and shrews from southern China. In this study, three particular viruses with zoonotic potential were molecularly examined in depth to investigate their relationship with human isolates.
-
Between 2013 and 2016, 225 rodents (Rattus norvegicus and Rattus flavipectus) and 196 shrews (Suncus murinus) were trapped by mouse traps, using fried foods as bait, in Shenzhen city, Guangdong province, southern China. Samples of various tissues (liver, lung, and intestine) were collected. Rodents and shrews were first identified based on morphology; the species of animals from which virus-positive samples had been collected were further confirmed by sequencing of the mitochondrial cytochrome b gene (Ge et al., 2013). The tissue samples were reserved at –80 °C until nucleic acid isolation.
-
Tissue samples were suspended at 10% w/v in 1 mL of phosphate-buffered saline (pH 7.4), and centrifuged at 12,000 × g for 10 minutes at 4 °C (Eppendorf, Hamburg, Germany). Total viral nucleic acids from liver, lung, and intestine samples were extracted from 200 μL of tissue suspension separately, using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Nucleic acids were aliquoted with elution buffer and stored at –80 °C until subsequent use.
-
First, we mixed RNA extracted from liver, lung, and intestine samples from each animal. RT-PCR assays were conducted for the distinct viruses, using the One-Step RT-PCR Kit (Invitrogen, Karlsruhe, Germany); sequences were amplified to perform phylogenetic analyses. Primer combinations targeting specific virus are listed in Table 1.
Virus Primer Sequence (5′–3′)a Region Amplicon size (bp) Reference Mammarenavirus Arena-F1 AYNGGNACNCCRTTNGC L gene First round: 938 Li et al., 2015 Arena-R1 TCHTAYAARGARCARGTDGGDGG Arena-F2 GGNACYTCHTCHCCCCANAC Second round: 611 Arena-R2 AGYAARTGGGGNCCNAYKATG Hantavirus HAN-L-F1 ATGTAYGTBAGTGCWGATGC L gene First round: 453 Klempa et al., 2006 HAN-L-R1 AACCADTCWGTYCCRTCATC HAN-L-F2 TGCWGATGCHACIAARTGGTC Second round: 385 HAN-L-R2 GCRTCRTCWGARTGRTGDGCAA HEV DE-F4228 ACYTTYTGTGCYYTITTTGGTCCITGGTT RdRp gene First round: 371 Drexler et al., 2012 DE-R4598 CCGGGTTCRCCIGAGTGTTTCTTCCA DE-R4565 GCCATGTTCCAGAYGGTGTTCCA Second round: 338 Note: aR: G/A; Y: C/T; S: G/C; W: A/T; M: A/C; K: G/T; H: A/C/T; N: A/T/C/G; I: inosine. Table 1. Sequences of primers used for RT-PCR and sequencing
For mammarenaviruses, samples were screened by nested RT-PCR with primers amplifying the L segment as described previously (Li et al., 2015). For hantaviruses, samples were screened by RT-PCR using consensus primers based on the conserved domain of the L fragment (Klempa et al., 2006). For HEV, a hemi-nested RT-PCR targeting the viral RNA dependent RNA polymerase (RdRp)gene was performed, as described previously (Drexler et al., 2012).
-
RT-PCR products were first examined by agarose gel electrophoresis. Bands of the expected length were purified using the QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany), and then cloned into the vector pGEM-T Easy (Promega, Madison, WI, USA). The inserts of plasmids were sequenced with primers M13F and M13R (Invitrogen, Karlsruhe, Germany), using a 3100 Sequencer (ABI, Waltham, MA, USA). Preliminary sequence data were analyzed using Geneious (Version 10.0.2, Biomatters Limited, Auckland, New Zealand) (Wang et al., 2017). The derived sequences were submitted to the GenBank database under the subsequent accession numbers: mammarenavirus (KY659332− KY659346), hantaviruses (KY659347−KY659375), and HEV (KY659376−KY659382). Sequence similarity comparisons were implemented using the BLASTn search engine (https://blast.ncbi.nlm.nih.gov). Sequence alignment with other representative viral sequences was performed using MAFFT (Katoh et al., 2002). The phylogenetic trees were built using the neighbor-joining algorithm with bootstrap values supported by 1,000 replications in MEGA version 7 (Kumar et al., 2016).
-
In total, 421 rodents and shrews were captured between 2013 and 2016 in 10 regions of Shenzhen city: Guangming, Longhua, Bao’an, Nanshan, Futian, Luohu, Longgang, Pingshan, Yantian, and Dapeng districts. Tissue samples harvested from these animals were examined by RT-PCR for the presence of mammarenavirus, hantavirus, and HEV. As illustrated in Table 2, the detection rates for these viruses varied: mammarenavirus infection rates in Rattus norvegicus and Suncus murinus were 6.70% and 0.51%, respectively, with an overall positive rate of 3.56% (15 of 421 animals). Regarding hantavirus, the infection rates in Rattus norvegicus and Suncus murinus were 13.40% and 0.51%, respectively, with an overall positive rate of 6.89% (29 of 421 animals); therefore, hantaviruses were the most prevalent viruses in this study. HEV was detected in 7/209 Rattus norvegicus samples (2.87%) and 1/196 Suncus murinus samples (0.51%), with an overall positive rate of 1.66% (7 of 421 animals). However, R. flavipectus was negative for all three viruses. Positive samples for the different viruses were derived from 8 of the 10 districts.
Virus Species Total R. norvegicus R. flavipectus S. murinus Mammarenavius a14/209 (6.70) 0/16 (0.00) 1/196 (0.51) 15/421 (3.56) Hantavirus 28/209 (13.40) 0/16 (0.00) 1/196 (0.51) 29/421 (6.89) HEV 6/209 (2.87) 0/16 (0.00) 1/196 (0.51) 7/421 (1.66) Note: apositive samples/tested samples (%) Table 2. Detection rates of mammarenavius, hantavirus and HEV by RT-PCR in small mammals derived from Shenzhen city, China
-
A 589-bp sequence of the L fragment of mammarenavirus, from 15 samples from 14 rodents and 1 shrew, was amplified and sequenced. The 15 partial L fragment sequences had 93.8%–100% nucleotide sequence identity. Similarity comparisons reflected the highest nucleotide sequence similarity of these strains from Shenzhen city with the Cardamones virus isolate C617 from Cambodia (90.2%), followed by the Wenzhou virus isolate Rn-366 from eastern China (89.5%). Comparison of these strains with Lassa virus (LASV) revealed a lower nucleotide sequence similarity of up to 74.5%. A phylogenetic tree was built from the amplified sequences together with representative mammarenavirus strains (Figure 1). As shown in the tree, 15 strains clustered within mammarenaviruses from the Old World, consisting of Africa, Europe, and Asia, which is clearly separated from the New World comprising the Americas.
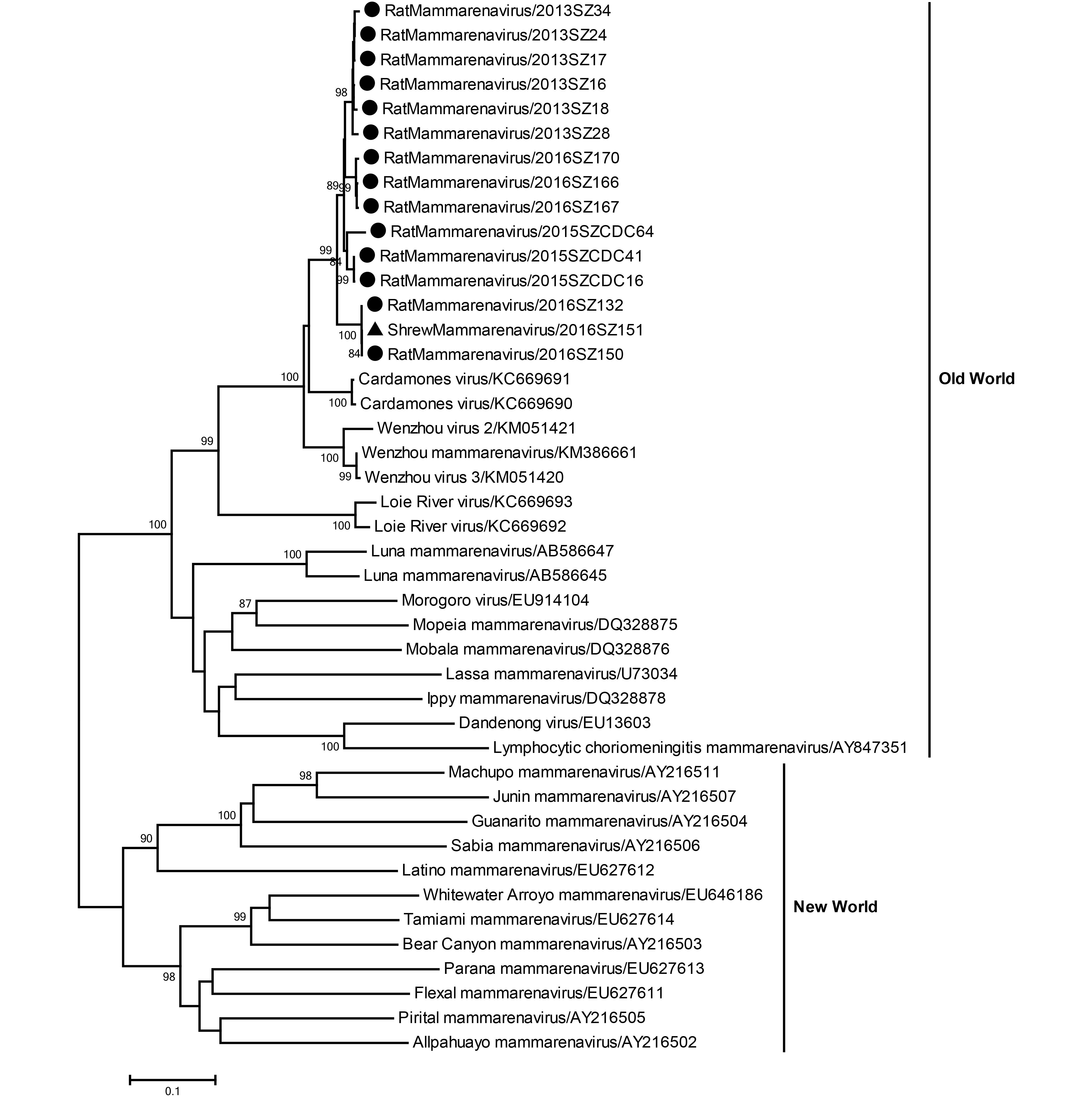
Figure 1. Phylogenetic relationships for 15 rodent and shrew mammarenavirus strains derived from Shenzhen city, China. The tree, which was based on a 589-bp sequence of the L fragment of the mammarenavirus, was built using a neighbor-joining algorithm implemented in Mega version 7. Bootstrap values (> 70%) are shown at specific nodes. Scale bars reflect the number of nucleotide substitutions per site. The tree is divided between the Old World and New World mammarenaviruses. The strain designations are reflected at each branch. The newly determined Shenzhen rodent and shrew strains are indicated via circles and triangles, respectively.
-
A 359-bp sequence of the L fragment of hantavirus was amplified and sequenced from 29 samples collected from 28 rodents and 1 shrew. The 28 partial L fragment sequences from rodents had 96.5% to 100% nucleotide sequence similarity; these strains reflected the highest nucleotide sequence identity with the Seoul virus isolate 2012–289 from Fujian province, eastern China (99.7%). A shrew strain had the highest nucleotide sequence similarity with the Thottapalayam virus strain VRC-66412 from shrews in India (88.9%), followed by the Imjin virus strain Cl 04-55 from Crocidura lasiura in South Korea (70.6%); these viruses are highly divergent from human strains. A phylogenetic tree was constructed using representative hantavirus strains (Figure 2). In this tree, 28 rodent strains clustered together with the human Seoul virus, and a shrew strain clustered with the shrew Thottapalayam virus.
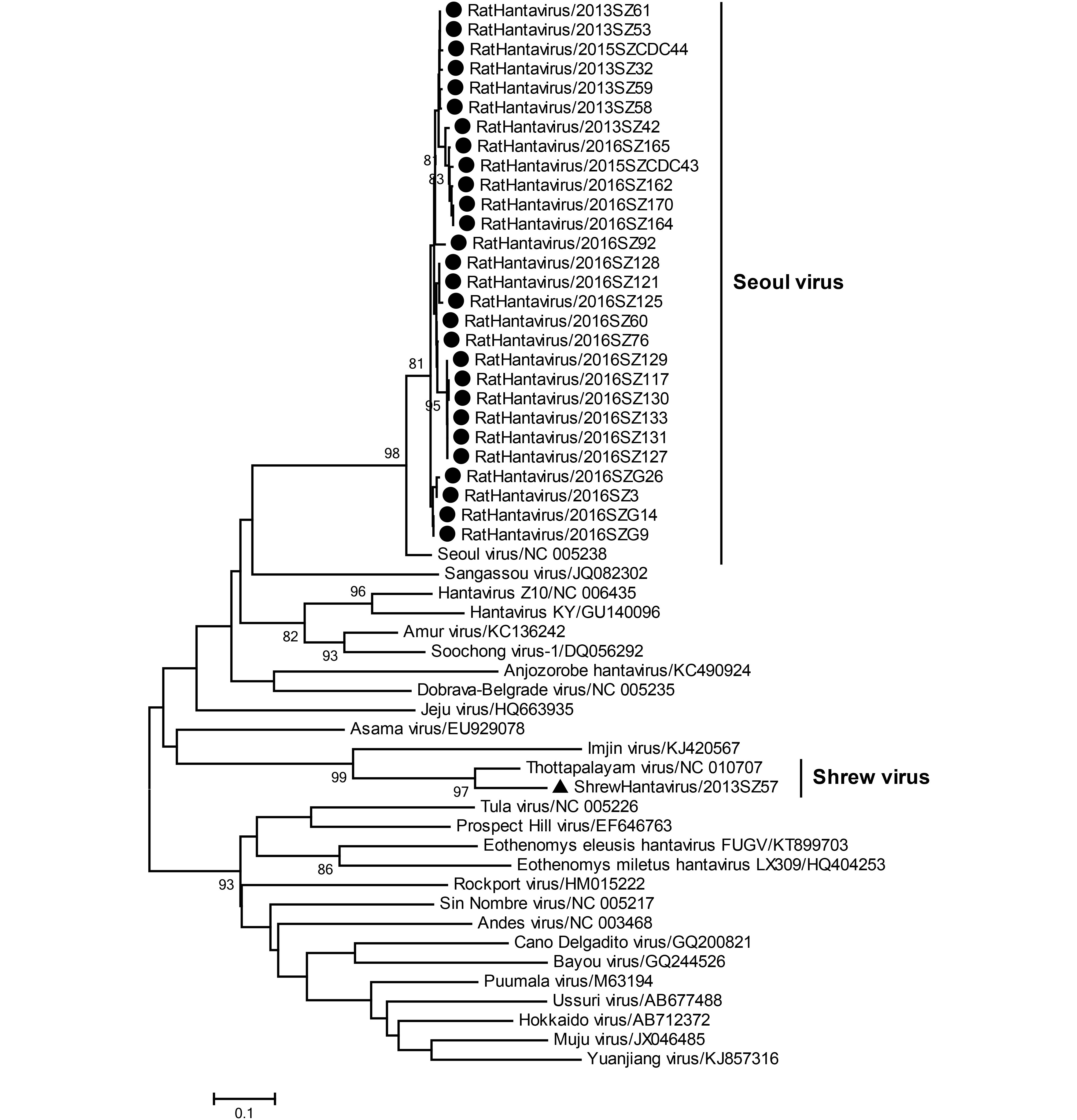
Figure 2. Phylogenetic relationships for 29 rodent and shrew hantavirus strains derived from Shenzhen city, China. The tree, which was based on a 359-bp sequence of the L fragment of the hantavirus, was built using a neighbor-joining algorithm implemented in Mega version 7. Bootstrap values (> 70%) are shown at specific nodes. Scale bars reflect the number of nucleotide substitutions per site. Seoul virus and Shrew virus are indicated on the right. The strain designations are indicated at each branch. The newly determined Shenzhen rodent and shrew strains are indicated via circles and triangles, respectively.
-
A 280-bp fragment of the ORF1 of HEV, from 7 samples from 6 rodents and 1 shrew, was amplified and se-quenced. The 7 partial ORF1 fragment sequences had 77.7%–99.7% nucleotide sequence identity. Among them, 4 HEV strains from 3 rodents and 1 shrew had the highest nucleotide sequence similarity of 84.7% with Hepatitis E virus rat/R63/DEU/2009 from rodents in Germany; the other 3 rodent HEV strains had the highest identity of 84.9% with Hepatitis E virus isolate Vietnam-105 from rats in Vietnam. A phylogenetic tree was constructed using representative HEV strains (Figure 3). According to the tree, all 8 strains from Shenzhen city belonged to species Orthohepevirus C (strains from rat, shrew, greater bandicoot, ferret, and mink). Of note, 3 rodent and 1 shrew HEV strains clustered together with rat HEV strains from Asia, whereas the other 3 rodent HEV strains clustered with rat strains from Europe.

Figure 3. Phylogenetic relationships for 7 rodent and shrew HEV strains derived from Shenzhen city, China. The tree, which was based on a 280-bp sequence of the RdRp gene of HEV, was constructed using a neighbor-joining algorithm implemented in Mega version 7. Bootstrap values (> 70%) are indicated at specific nodes. Scale bars indicate the number of nucleotide substitutions per site. The strain designations are reflected at each branch. The newly determined Shenzhen rodent and shrew strains are indicated via circles and triangles, respectively
-
Rodents are major reservoirs of human disease owing to their close contact with humans. In this study, we collected samples from rodents and shrews in Shenzhen, China, which has a high population density, and at-tempted to detect three viral families in these samples. All three viral families were detected, with infection rates of 3.56% (mammarenavirus), 6.89% (hantavirus), and 1.66% (rat HEV). Notably, positive samples for different viruses were derived from 8 of 10 districts where wild small mammals were captured. These results indicate that the detected viruses are widely distributed in Shenzhen. Further surveillance of interspecies transmission is needed to control the zoonotic spread of these diseases to human populations.
The only mammarenaviruses that have been isolated in China to date are LCMV (Morita et al., 1996) and WENV (Li et al., 2015). Recently, a genetic variant of WENV was shown to be associated with human infection in Southeastern Asia (Blasdell et al., 2016). The strains detected in this study showed high similarity with WENV and WENV-like viruses from Cambodia, suggesting a potential threat to human health. Notably, the mammarenaviruses found in rodents and shrews are almost identical based on their partial L-gene sequence. This result is consistent with previous findings in other regions, suggesting frequent interspecies transmission of this virus between rodents and shrews, which share common niches (Li et al., 2015).
HTNV and SEOV are the major causative agents of HFRS in China (Zhang et al., 2004). During the last decade, several cases of HFRS have been reported in Shenzhen city (Yang et al., 2006). Here, SEOVs were detected in rodents with a high infection rate, indicating that these SEOV strains are dominant strains associated with human diseases in this city. Hantaviruses have recently been detected in shrews in China; however, it is not clear whether this prevalence is associated with human illness (Guo et al., 2011; Zuo et al., 2014). Only one virus, which was similar to the Thottapalayam virus, was found in shrews.
After the discovery of animal HEV strains closely related to human HEV, the concept of zoonotic hepatitis E emerged (Pavio et al., 2015). Recently, rat HEV has been discovered in various species of rodents and shrews in China (Guan et al., 2013; Li et al., 2013). In this study, we found that rodents and shrews in Shenzhen city are infected with genetically diverse strains of rat HEV, which are similar to those found in both Asian and European rats. Similar rat HEVs were found in rodents and shrews, indicating interspecies transmission of these viruses between two different hosts (Guan et al., 2013). However, these rodent and shrew strains were divergent from human HEV. Therefore, their potential threat to humans needs to be further studied.
In conclusion, in the present work, several zoonotic viruses were detected in rodents and shrews from urban and rural districts of Shenzhen city, China; some of these strains showed significant zoonotic potential. To decrease the risk of infection in humans via contamination of food products and water, application of good hygiene practices aimed at minimizing contact with rodents and shrews is necessary. In further studies, important viruses that have already been found in rodents and shrews should be investigated for assessment of their risk of transmission to humans; for example, the relationship between rodent and human coronavirus (Tsoleridis et al., 2016), hepacivirus (Drexler et al., 2013), and astrovirus (Chu et al., 2010). To achieve a more accurate evaluation of the potential of zoonotic viruses from rodents and shrews, detection from greater sample numbers and wider geographical regions, as well as application of improved diagnostic methods, should be considered.
-
This work was jointly funded by the Scientific and Technological Basis Special Project (2013FY113500), the National Natural Science Foundation of China (Grant No. 81290341), and the China Mega-Project for Infectious Disease (2014ZX10004001-003) from the Minister of Science and Technology of the People’s Republic of China to ZL S and the Basic Research Project of Shenzhen Science and Technology Innovation Program (JCYJ2015 0402102519532) to CLCai. BW was supported by the China Scholarship Council (CSC), Beijing, China.
-
The authors declare that they have no conflict of interest. This study was approved by the Animal Ethics Committee of the Wuhan Institute of Virology (Animal ethics approval number: WIVA05201202). All institutional and national guidelines for the care and use of animals were followed.
-
XLY and ZLS designed and coordinated the study. CLC collected samples. XLY, CLC, BW, BL, WZ and YZ performed molecular studies. XLY and BW analyzed the data. BW, CLC, ZLS and XLY drafted the manuscript. All authors read and approved the final manuscript.
Detection and characterization of three zoonotic viruses in wild rodents and shrews from Shenzhen city, China
- Bo Wang 1,# ,
- Chun-Lin Cai 2,# ,
- Bei Li 1 ,
- Wei Zhang 1 ,
- Yan Zhu 1 ,
- Wei-Hong Chen 2 ,
- Fei Zhuo 2 ,
- Zheng-Li Shi 1 ,
-
Xing-Lou Yang
1,,

- Received Date: 10 March 2017
- Accepted Date: 27 May 2017
- Published Date: 17 July 2017
Abstract: Diverse species of rodents and shrews,which are abundant worldwide,harbor a variety of viruses; some of these are closely related to human viruses and possess zoonotic potential.Previously studies have demonstrated that the mammarenavirus and hantavirus carried by rodents or shrews could cause diseases in human population.To determine the distribution of zoonotic viruses in Shenzhen city,the major city in southern China with a high population density,we analyzed 225 rodents (Rattus norvegicus and Rattus flavipectus) and 196 shrews (Suncus murinus) from urban and rural districts for the presence of mammarenavirus,hantavirus,and hepatitis E virus (HEV) by RT-PCR targeting the conserved regions.The infection rates for mammarenavirus,hantaviruses, and HEV in rodents and shrews were 3.56%,6.89%,and 1.66%,respectively.Partial genome fragment analysis indicated that mammarenavirus and hantavirus strains had more than 90% and 99% nucleic acid identity with Cardamones virus and Seoul virus,respectively,which cause diseases in humans.Although the present HEV strains identified are typically found worldwide, phylogenetic analysis demonstrated a divergence of 16%.To our knowledge,the present work is the first report of the prevalence of mammarenavirus,hantaviruses,and rat HEV strains in rodents and shrews from Shenzhen city,China.Our findings highlight the zoonotic potential of rodent-and shrew-borne mammarenavirus and hantavirus,and the biodiversity of rat HEV isolates in Shenzhen city.The present work suggests that utilization of good hygiene habits is important to minimize the risk of zoonosis.

 DownLoad:
DownLoad: