












-
Dear Editor,
Porcine reproductive and respiratory syndrome virus (PRRSV), a member of the family Arteriviridae, represents one of the most challenging pathogens in the swine industry, with serious economic impact. Unfortunately, despite widespread application, vaccination has not been successful in effectively controlling the virus, mainly because of the limited cross-protection among different strains (Kimman et al., 2009).
A longitudinal study revealed a gradual rise in genetic divergence among field isolates of PRRSV type 2 (Brar et al., 2015). Similar to other RNA viruses, PRRSV displays high mutation and recombination rates, which give rise to a plethora of new variants able to rapidly explore the fitness landscape. The substantial viral population size at the host and the population level should create optimal conditions for natural selection to act. Accordingly, different studies have pointed out selective pressures, mainly related to host immunity, which affect PRRSV evolution (Franzo et al., 2015; Goldberg et al., 2003). However, the level at which these forces act is still a matter of debate. Although intra-host viral variability has been reported and escape mutants have been identified in experimental settings (Costers et al., 2010), their roles in viral evolution have been often downgraded (Brar et al., 2015).
A recent study based on deep sequencing evidenced that high quasispecies diversity occurs during the transition from the acute to the rebound stage of infection, and the introduction of mutations resulting in a gain of fitness could provide a mechanism of persistence, potentially increasing within-animal fitness (Chen et al., 2016). These variants could be transmitted to other pigs (Goldberg et al., 2003), leading to further evolution and spread of new variants. However, a trade-off between adaptation to specific individuals and transmissibility has been reported for some viruses (Pybus and Rambaut, 2009). In this study, a large-scale analysis of selective-pressure action was conducted to investigate whether intra-host adaptation and immuno-escape can be the source of population-level variability. Selective pressures acting at the population level on 5 regions of 4 different viral proteins (NSP1, NSP2, GP3, and GP5) were estimated and compared with those acting at the individual level in experimental conditions.
A collection of PRRSV type 2 full genomes was downloaded, and sequences related to Ingelvac PRRSV®MLV, Prime PacTM, and Ingelvac PRRSV®ATP vaccines were removed. The remaining genomes (i.e., 382 sequences) were aligned using the MAFFT method (Katoh and Standley, 2013), and regions in the 5 segments located in the 4 proteins in which positive selection was detected by Chen et al. (2016) were selected. The analysis was limited to these regions to allow proper comparison with experimental data (Chen et al., 2016) for robust evaluation of the forces acting at different levels of PRRSV evolution. Only complete genomes were included to guarantee consistency between sequence datasets. The sequences were aligned at the amino-acid level and then back-translated to nucleotide alignments using TranslatorX (Abascal et al., 2010). For each region, recombination breakpoints were identified using GARD (Kosakovsky et al., 2006), and for each non-recombinant partition, a phylogenetic tree was reconstructed using PhyML (Guindon et al., 2010).
Partition-specific trees were used to analyze selective pressures by the Single Likelihood Ancestor Counting (SLAC), Mixed Effects Model of Evolution (MEME), and Fast Unconstrained Bayesian AppRoximation (FUBAR) methods implemented in HyPhy (Kosakovsky Pond et al., 2005). Significance was set at P < 0.05 for MEME and at P < 0.1 for SLAC, which is more conservative. The results of FUBAR were accepted when the posterior probability was greater than 0.9. Sites were assumed to be under diversifying selection when detected by MEME, which can model episodic diversifying selection, or by at least two other methods. Positions detected to be under positive selection were mapped to the genome of the reference strain (NVSL97-7895; Acc. No: AY545985) used by Chen et al. (2016) . Per-site ancestral-sequence reconstruction was conducted on positions detected under statistically significant diversifying selection in both the current study and in Chen et al. (2016) , by using the maximum-likelihood approach of the ape package implemented in R (Paradis et al., 2004). The partition-specific phylogenetic trees were used for this purpose.
Compared to the results of Chen et al. (2016) , a greater number of positions within each region were identified to be under pervasive or episodic diversifying selection (Table 1, Supplementary Figure S1). This is clearly expected considering the different scales of the two studies. The limited number of pigs (i.e., 4) analyzed by Chen et al. (2016) surely prevented exploration of the full set of potentially advantageous mutations that were investigated more comprehensively in the current study. The fact that some sites under diversifying selection identified in Chen et al. (2016) were not identified in this study can be explained by several reasons. Firstly, a chance effect causing an apparent statistically significant excess of temporary mutations cannot be excluded, particularly in the case of a small sample size. Secondly, several non-synonymous mutations could have persisted for a certain time even if not beneficial to the virus, before being purged by natural selection. Interestingly, Chen et al. (2016) reported an overall decrease in the number of detected substitutions at 42 days post infection after a peak at day 28, supporting this hypothesis. Finally, as proposed for the human immunodeficiency virus, the adaptation to the specific host immune system could, in some circumstances, carry a penalty in terms of transmissibility, decreasing viral fitness in other animals and the population-level success (Pybus and Rambaut, 2009). Despite these inconsistencies between both studies, it is noteworthy that half of the sites under diversifying selection at the animal level were identified also at the population level. In particular, position 113 of nsp1, positions 30 and 143 of GP3, and position 32 of GP5 were detected in both studies (Table 1, Supplementary Figure S1). Remarkably, these proteins play important roles in conditioning the host immune response by serving as epitopes and interacting with the cellular machinery (Darwich et al., 2010). The evidence that the a single position can be prone to diversification at both the individual and population levels supports the idea that some of the changes favoring virus persistence can be efficiently transmitted to other animals and spread on a large scale, providing further opportunity for selective pressure to act. Consequently, these results, combined with those of Chen et al. (2016) , provide strong evidence that the variability generated during viral persistence within the host can have a pivotal role in PRRSV evolution, suggesting that the strategy used to persist in a single animal can be translated into a strategy to survive and spread at the population level. The high homogeneity of swine populations from a genetic and immunological perspective could favor this phenomenon by reducing the difference between individual and population fitness. In fact, even if advantageous mutations within a host have a cost in terms of transmission fitness because of increased specialization to the particular environment, the uniformity that characterizes modern livestock populations is likely to lower the impact of this limiting factor. Consequently, although the emergence of fitter variants can be assumed to be rare and conditions favorable to evolution are probably sporadic (e.g., rebound infection has been reported only in approximately 15% of infections; Chen et al., 2016), other epidemiological and managerial factors are probably creating an ideal condition for the maintenance and spread of these variants (Franzo et al., 2016).

Figure 1. Partition-specific phylogenetic trees reconstructed using the maximum-likelihood method implemented in PhyML. Partitions were defined according to Chen et al., 2016. For graphical reasons, only regions for which amino-acid positions were proven under statistically significant diversifying selection in both the current study and in Chen et al. (2016) are reported. For each of these positions, the amino acid (named according to IUPAC nomenclature) is represented as a color-coded circle located on the corresponding tree tip (i.e., the sampled viral sequence). Additionally, the results of an amino-acid ancestral-sequence reconstruction are plotted: ancestral nodes (i.e., states) are represented as pie charts whose slices are proportional to the feature (i.e., amino acid) likelihood in the specific ancestral protein.
Proteina Codonb SLAC dN-dS SLAC P-value MEME ω+ MEME P-value FUBAR dN-dS FUBAR Post.Pr. nsp1 Region1(387 bp) 75 –1.633 0.991 >100 0.039 –0.167 0.020 111 –0.91 0.899 86.227 0.018 –0.097 0.111 113 –0.737 0.855 90.103 0.001 –0.053 0.229 171 –0.246 0.708 >100 0.006 –0.005 0.427 nsp1 Region2(336 bp) 193 –0.404 0.951 16.022 0.041 –0.053 0.197 267 0.896 0.001 >100 0 0.288 0.998 287 –0.288 0.921 31.674 0.039 –0.048 0.212 nsp2 Region7(462 bp) 754 –0.212 1.000 74.104 0.028 –0.709 0.002 772 –0.097 0.89 >100 0.001 –0.157 0.131 780 0.109 0.064 5.502 0.036 0.473 0.946 785 0.103 0.099 >100 0.007 0.372 0.891 796 0.068 0.22 39.974 0.049 0.246 0.759 811 –0.024 0.728 >100 0.003 –0.199 0.197 824 –0.083 0.926 9.693 0.033 –0.128 0.283 840 0.115 0.011 >100 0.017 0.486 0.964 842 0.019 0.48 23.925 0.027 –0.15 0.214 944 –0.008 0.641 >100 0.024 –0.043 0.352 GP3 Region8(357 bp) 30 0.823 0.004 >100 0 0.227 0.999 41 0.236 0.198 >100 0.035 0.075 0.885 77 –2.024 1.000 11.314 0.041 –0.362 0.003 97 –0.224 0.941 94.368 0.032 –0.123 0.062 134 0.839 0.065 2.219 0.095 0.525 0.985 143 0.747 0.072 12.568 0.002 0.519 0.982 GP5 Region9(294 bp) 13 3.021 0.035 4.153 0.021 0.235 0.977 14 2.496 0.002 >100 0.002 0.29 0.996 15 2.169 0.030 7.506 0.022 0.157 0.958 24 1.765 0.047 >100 0.005 0.177 0.981 25 3.102 0.008 16.273 0.002 0.361 0.995 26 2.400 0.008 >100 0.003 0.194 0.992 To be continued Table 1. Sites under statistically significant diversifying selection as determined by SLAC, MEME, and FUBAR
Despite these indicative results, this study has some limitations, mostly related to paucity of currently available data. Particularly, the complete genome sequences included in the current study were mainly from highly pathogenic strains from China and the U.S., and cannot be considered representative of the entire PRRSV type 2 population. Clearly, more extensive sampling, including asymptomatic animals, would greatly benefit the study of PRRSV evolution, and consequently, our understanding thereof. Nevertheless, despite these limitations imposed by practical and economical constraints, the use of a mathematical method able to detect sites under episodic diversifying selection (i.e., affecting even a single branch of the gene tree) substantially compensates for the effect of these biases. Additionally, although the viral population herein cannot be regarded as representative of the entire PRRSV type 2 population, the biological relevance of the results is not precluded. In fact, interactions between intra- and inter-host evolution have been proven to happen in the vast viral populations affecting developed (U.S.) and developing (China) countries, which are also relevant from a sanitary and economic point of view. Despite the lack of extensive data on intra-host viral evolution hampering the formulation of a definitive hypothesis, the present work, extending the findings of Chen et al. (2016) study to an epidemiological scale, provides a comprehensive, albeit preliminary, model linking the different levels of PRRSV evolution.
In summary, the present study demonstrates that the forces favoring intra-animal diversification can cause the emergence of variants that, benefiting of their diversity at the population level, are able to succeed and spread in an often highly homogeneous environment, and to be further selected later.
HTML
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors. Supplementary figure S1 is available on the websites of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-
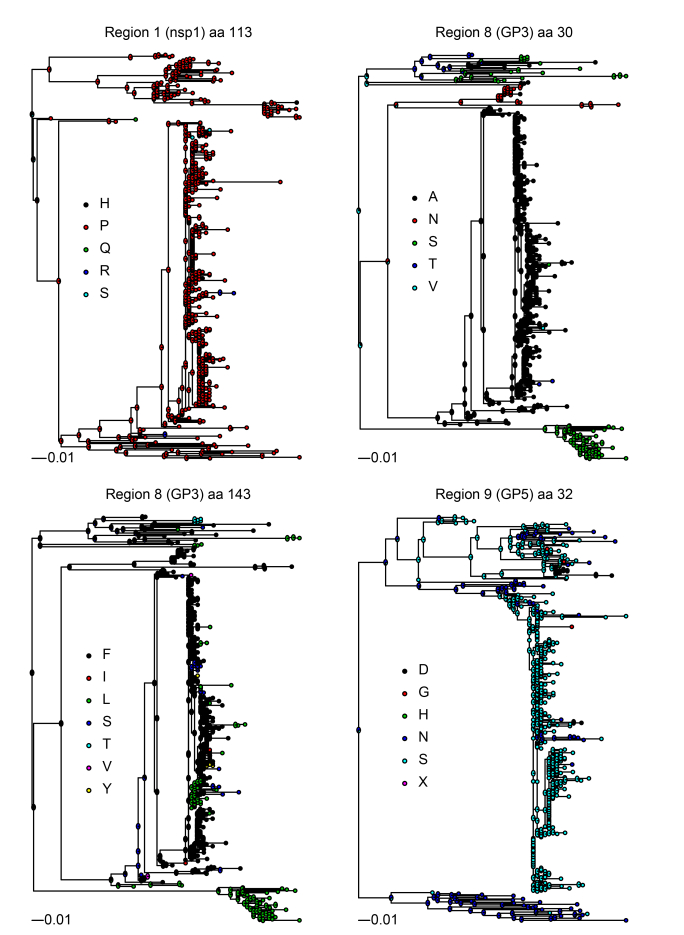
Figure S1. Partition-specific phylogenetic trees reconstructed using the maximum-likelihood method implemented in PhyML. Partitions were defined according to Chen et al., 2016. For graphical reasons, only regions for which amino-acid positions were proven under statistically significant diversifying selection in both the current study and in Chen et al. (2016) are reported. For each of these positions, the amino acid (named according to IUPAC nomenclature) is represented as a color-coded circle located on the corresponding tree tip (i.e., the sampled viral sequence). Additionally, the results of an amino-acid ancestral-sequence reconstruction are plotted: ancestral nodes (i.e., states) are represented as pie charts whose slices are proportional to the feature (i.e., amino acid) likelihood in the specific ancestral protein

 DownLoad:
DownLoad: