












HTML
-
According to a Joint United Nations Programme on HIV/ AIDS (UNAIDS) report, the number of people living with AIDS reached 36.7 million worldwide at the end of 2016. In addition of causing immune system dysfunction, human immunodeficiency virus (HIV) infection can lead to HIVassociated neurocognitive disorders (HAND), which are highly prevalent in HIV-infected individuals and severely affects the quality of their lives (Ellis et al. 2007; Robertson et al. 2014; Sacktor et al. 2016). HIV proteins (gp120, Tat, Vpr and Nef) can cause neuronal damage by interacting with neuron surface receptors or activating caspases (Rao et al. 2014). HIV transactivator of transcription (Tat) mRNA and the toxic Tat protein have been detected in the central nervous system (CNS) of patients with HAND (Shin and Thayer 2013). In HIV-1 Tat transgenic mice and cultured primary neurons, exposure to Tat1–86 markedly reduces the number of dendritic spines, which results in synapse loss and impaired cognitive function (Fitting et al. 2010, 2013).
Neurotrophins are involved in the regulation of synaptic plasticity and cognitive function. As a member of the family of secreted neurotrophins, brain-derived neurotrophic factor (BDNF) is a key regulator of synapse formation and plasticity that is widely expressed in the brain (Lu et al. 2013). Its expression is reduced in many neurodegenerative diseases, including Huntington's disease and Alzheimer's disease (Park and Poo 2013). In the adult brain, the main roles of BDNF are the enhancement of synaptic transmission, control of synaptic plasticity, and promotion of synaptic growth (Lu et al. 2013). Clinical experiments have shown that all areas of the brain in patients with HAND expressed significantly lower levels of BDNF compared to those in people with HIV but without dementia (Bachis et al. 2012). Moreover, HIV-1 dramatically decreased BDNF mRNA levels in T cells (Avdoshina et al. 2011). In addition, cAMP response element binding protein (CREB), which is a transcription factor, helps modulate synaptic plasticity (Benito and Barco 2010). CREB can interact with calcium/cyclic AMP response element (CRE) in the 5' proximal region of the BDNF promoter to induce its transcription. Phosphorylation of CREB at Ser133 is indispensable to transcriptional activation (Tao et al. 1998).
Protein serine/threonine phosphatase 1 (PP1) is one of the few phosphatases known to dephosphorylate CREB at Ser133 and inhibit CRE-mediated gene transcription (Alberts et al. 1994). It is highly enriched in human neurons and is involved in a wide range of cellular physiological processes, including regulation of cell membrane receptors and corresponding channels (Shi 2009). There are four variants of PP1: PP1α, PP1β, PP1γ1, and PP1γ2. Among these isoforms, PP1α and PP1γ1 are more frequently found in dendritic spines, whereas PP1β is predominately located in dendritic shafts (Bordelon et al. 2005). The activity of PP1 isoforms can be modulated by phosphorylation of a conserved C-terminal threonine residue (Dohadwala et al. 1994). With PP1α, cyclin-dependent protein kinase 2 (CDK2) can phosphorylate PP1 at Thr320 and inhibit PP1 activity in mitotic cells, which is necessary for the initiation of normal cell division (Kwon et al. 1997). Moreover, in cortical neurons, PP1 can be phosphorylated by CDK5 at Thr320, which suppresses N-methyl-D-aspartate receptor (NMDAR) activation (Hou et al. 2013). In addition, PP1 activity can be controlled by several regulatory proteins, including inhibitor-1 (I-1), I-2, and dopamine- and cAMP-regulated phosphoprotein-32 (DARPP-32) (Shi 2009). Among these three modulatory proteins, I-2 can suppress PP1 phosphorylation at Thr320 and positively regulate PP1 activity to constrain memory formation (Yang et al. 2015). On the other hand, the catalytic subunit of PP1 is capable of autodephosphorylation. Although PP1 has been found to modulate synaptic plasticity and memory formation, little is known about the effect of Tat exposure on PP1 and the CREB/BDNF signaling pathway during the induction of neurotoxicity.
This report describes a study focusing on whether PP1 is associated with Tat1–72-induced neurotoxicity in cultures of primary neurons. First, we performed neuron metabolic activity assay and immunofluorescence experiment to observe the effect of recombinant Tat1–72 on primary neurons. Next, changes of total PP1 and activated PP1 after exposure to Tat1–72 were analyzed by immunoblotting. Finally, we monitored CREB activity and CREB-mediated gene expression.
-
Tat (1–72 aa) was synthesized by DG Peptides Co., Ltd. (China) and reconstituted in sterile deionized water. The concentration of Tat's stock solution is 1 mmol/L. An aliquot of Tat protein was heat-inactivated by incubation at 85 ℃ for 30 min as previously described (Wayman et al. 2015).
-
Tat (3 μmol/L) was added to TZM-bl cells for 6 and 12 h. Then, the culture medium was removed, cells were gently washed with PBS, 100 μL of Glo lysis buffer was added, and the cells were incubated again for 5 min. Fifty microliters of lysate was mixed with 50 μL of Steady-Glo luciferase buffer to measure luciferase activity using the GloMax system (Promega).
-
Primary cortical neurons from C57BL/6 mice were prepared as described previously (Zhou et al. 2014). Pregnant mice (embryonic stages E17–18) were euthanized by dislocation and cleaned with 75% ethanol. Their uteruses were opened, and pups were placed in sterile pre-chilled phosphate-buffered saline (PBS). The brains of pups were placed on a stereoscope, and cortices were removed. After papain (Sigma, Germany) and deoxyribonuclease Ⅰ (Sigma, Germany) digestion, cortical neurons were counted and plated on a sterilized cover glass or 24-well plates in neurobasal medium (Gibco, USA) supplemented with 5% B27 (Gibco, USA). The medium was changed every 2–3 days. All experiments were performed with 10- to 12-day-old cultures, at which time the cultures contain 90%–95% neurons and 5%–10% astrocytes.
-
The metabolic activity of neurons was evaluated using a CCK-8 kit (BestBio, China) in accordance with the manufacturer's instructions. Briefly, 5000 cells were seeded in 96-well plates. After 10 days, various concentrations of Tat were added to the plate. After 24 h of incubation, CCK-8 solution (10 μL) was added, and the plates were incubated at 37 ℃ for an additional 4 h. The absorbance at 450 nm was measured using a microplate reader.
-
After fixation with 4% paraformaldehyde, the cultured neurons were blocked with PBS containing 5% horse serum and 0.1% Triton-X100 for 1 h at room temperature. The neurons were then incubated with anti-Class Ⅲ β-tubulin antibody (1:500 dilution; Abcam, USA) at 4 ℃ overnight. After washing with PBS with Tween 20 (PBST), the neurons were incubated with Goat Anti-Rabbit IgG H & L (TRITC) secondary antibody (1:1000 dilution; Abcam, USA) and incubated for 1 h at room temperature. Then, the samples were washed with PBS three times for 5 min each. All images were acquired using a 40× objective with a computer controlled microscope (Olympics, Japan). The total length of the dendrite for a single neuron was estimated using Image-Pro Plus software. All neurons that had clearly defined cell bodies located completely within the viewing field were included, whereas neurons connected to other neurons or neurite endings outside of the optic field were excluded. Fifty neurons were sampled per treatment group in each experiment. Data are presented as the mean dendritic length per neuron ± standard error of the mean (SEM) from at least three independent experiments.
-
A phosphoprotein extraction kit (BestBio, China) was used to extract neuronal proteins. The lysates were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to a polyvinylidene fluoride membrane, and then membranes incubated overnight at 4 ℃ with the corresponding primary antibody, namely anti-PP1 antibody (1:1000 dilution, Santa Cruz, USA), antiphospho-PP1α (Thr320) antibody (1:1000 dilution; Cell Signaling, USA), anti-CREB antibody (1:1000 dilution; Abcam, USA), anti-phospho-CREB (Ser133) antibody (1:1000 dilution; Abcam, USA), or anti-β-actin antibody (1:5000 dilution; Santa Cruz, USA). Then, the amounts of protein were determined using a chemiluminescent kit (Thermo Scientific, USA).
-
Total RNA was extracted from primary neurons using TRIzol reagent (Invitrogen) according to the instructions. Next, cDNA was synthesized using SMARTer Kits (Takara), and the cDNA was amplified using TransStart Green qPCR SuperMix (Transgen Biotech). The following primers were used: I-2: (forward primer: 5'-CCTGCTCTGATTTCC TAC-3' reverse primer: 5'-CAGCAGATGACAGACTAC- 3'), BDNF: (forward primer: 5'-GCCCAACGAAGAAAAC CATAA-3', reverse primer: 5'-GGAGGCTCCAAAGGCA CTT-3'), c-fos: (forward primer: 5'-CCGAAGGGAACG GAATAAGA-3', reverse primer: 5'-TCTGGGAAGCCA AGGTCAT-3'), Egr-1: (forward primer: 5'-GAAGGCGA TGGTGGAGACG-3', reverse primer: 5'-CGCAGCCGAG TATGGGAC-3') GAPDH: (forward primer: 5'-GTAG GAAGAGAGGGAAGAG-3', reverse primer: 5'-GAAGT CACAGGAGACAAC-3').
-
The data are shown as the mean ± SEM from at least three independent experiments. The Student's t test was used for the statistical analyses. P < 0.05 was considered statistically significant.
Tat1–72 Synthesis
Luciferase Reporter Assay
Cortical Primary Neuronal Culture and Tat Treatment
Metabolic Activity of Neurons
Immunofluorescence Stain
Western Blotting
Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
Statistical Analysis
-
Tat protein varies in length from 86 to 101 amino acids and is formed from two exons. The first exon contributes the first 72 amino acids and Tat1–72 has been reported to induce neuronal apoptosis (Eugenin et al. 2003; Maragos et al. 2003; Nath et al. 2012). Tat31–61 is responsible for most of the neurotoxic properties of the protein, with the neurotoxic epitope residing in this region (Nath et al. 1996). Moreover, the cysteine-rich domain of the first exon has been reported to be critical for dendritic pruning and synaptic loss in vitro (Bertrand et al. 2013). Hence, we utilized recombinant Tat1–72 in these studies.
To investigate the biological activity of recombinant Tat1–72 protein, luciferase activity was measured after the addition 3 μmol/L Tat to HIV-1 LTR-integrated TZM-bl cells. Figure 1A shows that 3 μmol/L Tat promoted LTR activity at 6 h when compared with the untreated group. Our result is in line with that of a report by Green and colleagues, who suggested that Tat protein is taken up by cells and that it rapidly stimulates HIV-1 transcription (Green and Loewenstein 1988). However, HIV-1 LTR transactivation decreased when peptide was added for 12 h. It seems highly probable that Tat is degraded by diverse mechanisms associated with autophagy or proteasomal or lysosomal degradation (Sagnier et al. 2015; Ali and Banerjea 2016).
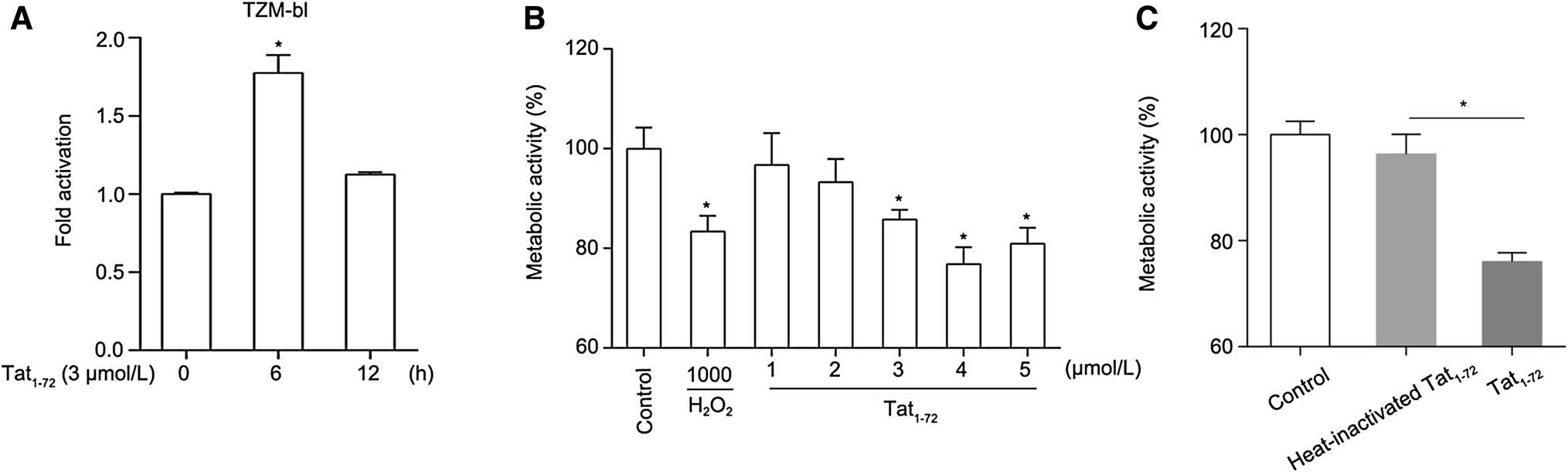
Figure 1. Recombinant Tat1–72 induces neurotoxicity in primary cultured neurons. (A) Recombinant Tat promoted HIV-1 LTR activation. Tat1–72 (3 μmol/L) was added to TZM-bl cells for 6 or 12 h, cells were lysed, and luciferase was detected using a GloMax system. Three replicates were sampled per treatment group in each of three independent experiments. Values are presented as mean ± SEM. *P < 0.05 versus 0 h group. (B) Tat decreased primary neuronal metabolic activity. Cultured neurons were treated with 1 mmol/L H2O2 or various concentrations of Tat1–72 (1, 2, 3, 4, or 5 μmol/L) for 24 h. H2O (5 μL) used as a control. Then, cell metabolic activity was detected using a CCK-8 kit. Five replicates were sampled per treatment group in each of three independent experiments. Values are presented as mean ± SEM. n = 3, *P < 0.05 versus control group. (C) Cultured neurons were treated with 5 μmol/L Tat1–72 or 5 μmol/L heat-inactivated Tat1–72 for 24 h. H2O (5 μL) used as a control. Then, cell metabolic activity was detected using a CCK-8 kit. Five replicates were sampled per treatment group in each of three independent experiments. Values are presented as mean ± SEM. n = 3, *P < 0.05 versus the control group
HIV-1 Tat is known to be toxic to neurons. In the present study, we evaluated the cytotoxicity of Tat in primary mouse cortical neurons. To investigate Tat-regulated neuronal metabolic activity, cultured neurons were treated with 1 mmol/L H2O2 or Tat at various concentrations (1, 2, 3, 4, or 5 μmol/L) for 24 h. By performing a CCK-8 assay, we found that cell metabolic activity declined dramatically with a Tat concentration of 3 μmol/L. H2O2, which causes cell death, was used as a positive control. Figure 1B shows that the toxic effects of 3 μmol/L Tat were nearly equivalent to those of H2O2. Our data suggested that the Tat1–72 could attenuate the metabolic activity of neurons, which is consistent with results described in previous reports that used Tat1–86 or Tat1–72 (Eugenin et al. 2007; Nath et al. 2012). In addition, we used 5 μmol/L heat-inactivated Tat1–72 as a negative control in the cell metabolic activity assay. The results showed that, compared to 5 μmol/L heat-inactivated Tat, 5 μmol/L Tat decreased neuronal metabolic activity (Fig. 1C).
-
To further assess the neurotoxicity of Tat in primary neurons, we treated neurons with different concentrations of recombinant protein (0, 1, 1.5, or 2 μmol/L) for 24 h and then stained them with anti-Class Ⅲ b-tubulin antibody and analyzed neuronal outgrowth. As shown in Fig. 2A, 2 μmol/L Tat significantly decreased the total length, thickness, and number of dendrites compared to those in the untreated group. Moreover, we determined the total length and number of dendrites of 50 neurons in each group using Image-Pro Plus software. Our data indicated that the number of dendrites per neuron decreased by 40% and that the total dendritic length per cell reduced by 25% (Fig. 2B and 2C). These findings suggested that Tat1–72 led to dendrite injury associated with Tat-induced neurotoxicity, which agrees with results described in previous reports which used Tat1–86 or Tat1–72 (Kim et al. 2008; Lu et al. 2011).
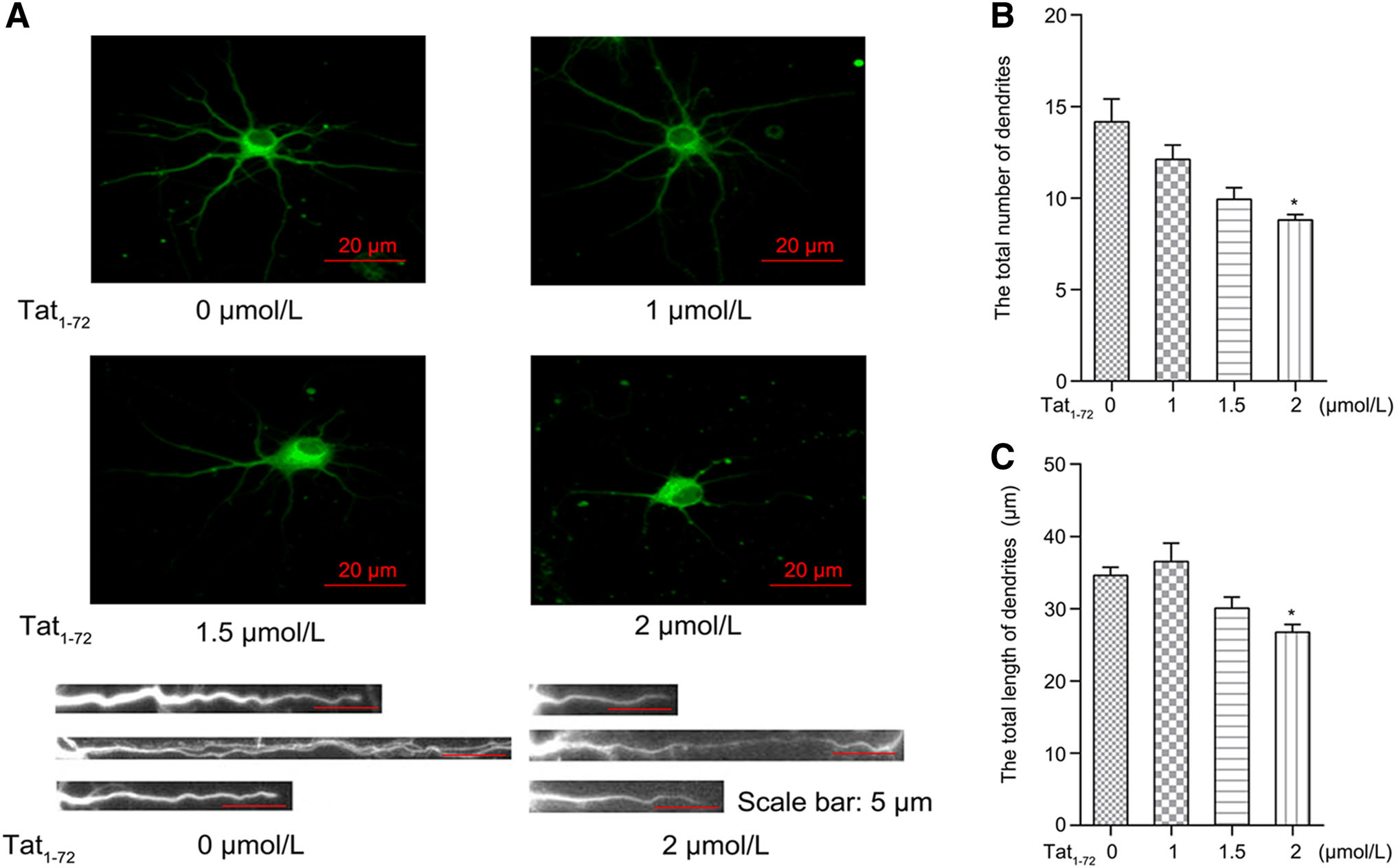
Figure 2. Exposure to Tat1–72 reduces the length and number of dendrites. (A) Primary cultured neurons were treated with various concentrations of Tat1–72 (0, 1, 1.5, or 2 μmol/L) for 24 h. Images were obtained by fluorescence microscopy (top). Enlarged view of individual dendritic branches (bottom). (B) Bar graph of the relative dendritic number per neuronal cell. All data are from three independent experiments. Values are presented as mean ± SEM. n = 3, *P < 0.05 versus the control group. (C) Bar graph of the relative total dendritic length per neuronal cell. H2O (2.5 μL) used as a control. All data are from three independent experiments. Values are presented as mean ± SEM. n = 3, *P < 0.05 versus the control group
-
In light of the Tat-induced loss of dendrites, we next asked what happened to PP1α, because nearly 70% of the isoform is exclusively located in dendritic spines (Munton et al. 2004). We investigated the levels of PP1 expression in lysates of neurons exposed (0, 0.5, 1, 2, 4, or 6 h) to Tat1–72 (2 μmol/L) by immunoblotting. As shown in Fig. 3A, treatment with Tat initially stimulated the expression of PP1, but, after 6 h of Tat exposure, the amount of PP1 decreased. Tat quickly induced the expression of PP1 in neurons, but this induction of PP1 expression was shortlived (Fig. 3B). This might be related to the regulation of enzyme expression by neurons themselves. In addition, the activity of PP1α is regulated by phosphorylation of Thr320 in its catalytic subunit (Dohadwala et al. 1994; Hou et al. 2013). Figure 3A and C show that Tat decreased phosphorylation of PP1 in a time-dependent manner. Phosphorylation of PP1 at Thr320, which was consistently strong after 0.5 h, weakened 1 h after treatment with Tat and was markedly attenuated after 6 h, suggesting that Tat can increase PP1 activity.

Figure 3. Tat1–72 modulates PP1 expression and activity. (A) Primary cultured neurons were treated with 2 μmol/L Tat1–72 for the indicated time. Then, the cells were lysed, and the lysate was subjected to western blotting. (B) PP1 expression was quantitatively analyzed. All data are from three independent experiments. Values are presented as mean ± SEM. *P < 0.05 versus 0 h group. (C) p-PP1 expression was quantitatively analyzed. All data are from three independent experiments. Values are presented as mean ± SEM. *P < 0.05 versus 0 h group. (D) Real-time PCR analysis of I-2 expression (relative to GAPDH expression) in cultured neurons treated with Tat1–72 (2 μmol/L) for 6 h. H2O (2.5 μL) used as a control. All data are from three independent experiments. Values are presented as mean ± SEM. **P < 0.01 versus the control group
Because I-2 positively regulates PP1 activity, we determined mRNA levels of I-2 in cultured neurons exposed to Tat (Sakamoto et al. 2011; Bennett and Lagopoulos 2014). As can be seen in Fig. 3D, 6 h of incubation with Tat highly upregulated I-2 mRNA levels. These results suggest that Tat1–72 affects synaptic plasticity, which is consistent with results in a previous report which used recombinant full-length Tat (Chang et al. 2011).
-
Because CREB is the substrate of PP1, we investigated the activity and expression of CREB by western blotting. As shown in Fig. 4A, the level of CREB phosphorylated at Ser133 was reduced by about 50% when neurons were treated with Tat for 24 h, compared to that when neurons were untreated, whereas the expression of CREB was not significantly different between the two groups. In addition, as determined by RT-qPCR assay, Tat exposure for 6 h markedly decreased the expression of CREB mRNA compared to that in the untreated group, suggesting that both the expression and activity of CREB are decreased by treatment with Tat (Fig. 4B). We also examined BDNF, cfos, and Egr1 mRNA levels by RT-qPCR following treatment with Tat (2 μmol/L) for 24 h. Figure 4C shows that the expression of these three genes decreased by about 50%. This finding suggested that Tat1–72 exposure decreases the expression of pCREB and inhibits the expression of CREB-regulated genes.
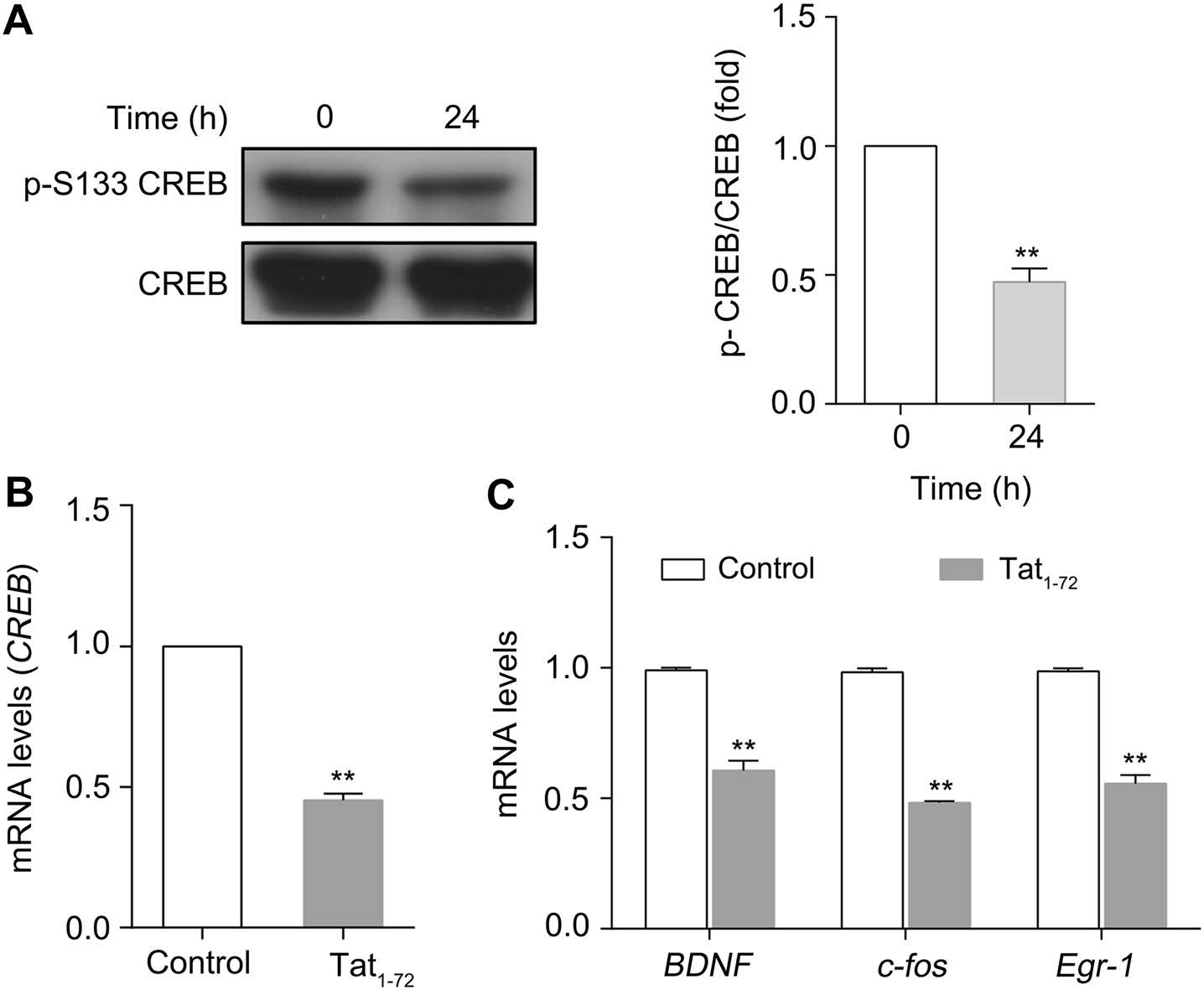
Figure 4. Tat1–72 downregulates CREB activity and CREB-mediated gene expression. (A) Primary cultured neurons were treated with 2 μmol/L Tat1–72 for 24 h. Then, the cells were lysed, and the lysate was subjected to western blotting. All data are from three independent experiments. Values are presented as mean ± SEM. **P < 0.01 versus 0 h group. (B) Real-time PCR analysis of CREB expression (relative to GAPDH expression) in cultured neurons treated with Tat1–72 (2 μmol/L) for 6 h. H2O (2.5 μL) used as a control. All data are from three independent experiments. Values are presented as mean ± SEM. **P < 0.01 versus the control group. (C) Real-time PCR analysis of BDNF, c-fos, and Egr-1 expression (relative to GAPDH expression) in cultured neurons treated with Tat1–72 (2 μmol/ L) for 24 h. H2O (2.5 μL) used as a control. All data are from three independent experiments. Values are presented as mean ± SEM. **P < 0.01 versus the control group
Recombinant Tat1–72 Induces Neurotoxicity in Primary Cultured Neurons
Exposure to Tat1–72 Reduces the Length and Number of Dendrites
Tat1–72 Modulates PP1 Expression and Activity
Tat1–72 Downregulates the Expression of Phosphorylated CREB and CREB-Mediated Gene Expression
-
The prevalence of neurocognitive impairment is increasing in patients with AIDS, even in the era of combined antiretroviral therapy. In early stages, patients with HAND have difficulties with attention and memory, suggesting neuronal injury (Eggers et al. 2017). HIV-1 Tat protein changes the structure of dendrites, which impairs learning and memory in mice (Genoux et al. 2002; Fitting et al. 2013). In this study, we initially demonstrated the neurotoxicity of recombinant Tat1–72 in primary cultured cortical neurons. Subsequently, we found that the number and length of neuronal dendrites decreased with exposure to Tat1–72. Moreover, we observed that the level of PP1 was significantly increased when neurons were treated with Tat1–72 for 2 h, but the PP1 level returned to baseline with 6 h of Tat1–72 treatment. Tat1–72 also activated PP1 by decreasing the level of PP1 phosphorylation at Thr320. Finally, we observed that both the levels of CREB phosphorylated at Ser133 and mRNA of BDNF, which is the target gene of CREB, dramatically declined.
PP1 is a ubiquitously expressed protein phosphatase that controls various cellular functions, including transcription, protein synthesis, glycogen metabolism, apoptosis, and cellular division (Rebelo et al., 2015). In the brain, PP1 was found to suppress learning and memory by decreasing the phosphorylation of its targets, including Ca2+/ calmodulin-dependent protein kinase Ⅱ (CaMKII), the GluA1 subunit of the alpha-amino-3-hydroxy-5-methyl-4- isoxazolepropionic acid (AMPA) receptor and GluN2B subunit of N-methyl-D-aspartate (NMDA) receptor (Genoux et al. 2002). In postsynaptic densities, CaMKII Thr286 can be dephosphorylated by PP1, which decreases CaMKII activity and long-term potentiation (LTP) (Strack et al. 1997). Moreover, neurabin-1 anchors PP1 through RVXF modular binding motifs and recruits it to synapses. Then, neurabin-PP1 complex promotes dephosphorylation of the AMPA receptor at GluA1 Ser831 and Ser845 during long-term depression (Hu et al. 2007). In addition, PP1 can regulate NMDAR activation by dephosphorylating Ser1303 in the GluN2B subunit, which decreases NMDAR activity and attenuates excitotoxicity (Dore et al. 2015). Our data showed that PP1 activity was elevated after exposure to Tat for 6 h, suggesting that Tat might limit the capacity of learning and memory in HAND patients through increased PP1 activity. Moreover, our data revealed that I-2 mRNA levels increased after cultured neurons were exposed to Tat for 6 h, suggesting that I-2 might cooperate with PP1 to promote dephosphorylation of PP1 target genes and control memory formation during Tat exposure.
CREB acts as a transcription factor. Phosphorylation of CREB at Ser133 is critical for CREB-dependent transcription, but CREB can be dephosphorylated by PP1, which results in the loss of its transcriptional activation capacity. Zhong et al. reported that Tat promoted CREB phosphorylation in hCMEC/D3 cultures (Zhong et al. 2012). In contrast, Zhu and colleagues found that exposure to Tat attenuated phosphorylation of CREB in rat nucleus accumbens (Zhu et al. 2015). Here, we have shown that phosphorylation of CREB at Ser133 decreased when Tat was administered for 24 h to cultured primary mice neurons, suggesting that Tat modulated the phosphorylation status of CREB Ser133, possibly through different cell signaling pathways in neurons and endothelial cells. Our results also suggest that PP1 activation might attenuate CREB phosphorylation following exposure to Tat and provide convincing evidence that HIV-1 Tat inhibits learning and memory.
BDNF plays a critical role in synaptic plasticity, and deletion of the BDNF gene significantly decreases LTP. Our results showed a * 50% decrease in BDNF mRNA levels when neurons were exposed to Tat for 24 h, suggesting that HIV-1 Tat protein, in addition to gp120, can reduce the expression of this neurotrophin and attenuate LTP by suppressing BDNF gene expression. CREB, phosphorylated at Ser133, can bind to the BDNF promoter to activate the transcription of the secreted protein. Our experiments showed that Tat decreased the phosphorylation of CREB and BDNF mRNA levels, indicating that HIV-1 Tat might downregulate BDNF mRNA levels by suppressing phosphorylation of CREB.
Dendrites are the main neuronal structure, and they receive and compute information from other cells. Moreover, proper dendrite morphology plays a crucial role in normal nervous system function (Martinez-Cerdeno 2017). Consistent with previous reports indicating that HIV-1 gp120 and Tat can impair dendrites, our data show that Tat significantly decreased the total length and number of dendrites per cell, suggesting that Tat-mediated neurotoxicity is the result of dendrite damage (Garden et al. 2002; Deyer et al. 2015). Dendritic spines contain neurotransmitters and proteins that participate in synaptic transmission and regulate synaptic plasticity; therefore, functions such as learning and memory depend on the stability of dendritic spines (Koleske 2013). PP1 isoforms such as PP1α and PP1γ1 are located in dendritic spines, and they modulate AMPA and NMDA receptor activation. Our results revealed that PP1 expression and activity changed following treatment with HIV-1 Tat, suggesting that Tat might impair dendritic spines and affect synaptic plasticity by changing PP1 expression and activity. In addition, BDNF plays a key role in dendritic spine stability because BDNF induces an increase in F-actin in spines, and F-actin changes spine structure and maintains spine stability (Rex et al. 2007; Koleske 2013). Our findings indicate that exposure to Tat can inhibit BNDF transcription and disrupt the stability of dendrites and spines.
In view of the cysteine-rich domain of the first exon is critical for dendritic pruning and synaptic loss, so we utilized recombinant Tat1–72 in our studies. While, in this paper we used 2 μmol/L Tat1–72 which is much higher than its physiological concentration which are approximately 40 ng/mL (~10 nmol/L) (Xiao et al. 2000). There are two main reasons for using high concentration of Tat1–72 in this study, one is the recombinant Tat is oxidized or denatured during the purification process (Li et al. 2008), another is the uptake of the first exon (Tat1–72) has been reported to be less efficient than that of extracellular Tat1–86 (Ma and Nath 1997).
In conclusion, our data show that HIV-1 Tat1–72 can promote PP1 activity while suppressing CREB activity and BDNF expression, suggesting that these proteins are involved in dendrite and spine injury. Moreover, our study provides new evidence for Tat-mediated neurotoxicity. However, we have limited information on the relationship between PP1 and BDNF with Tat exposure. The detailed mechanism underlying Tat control of synaptic plasticity must be determined in future studies.
-
This work was supported by Grants from the National Natural Science Foundation of China (81571987).
-
YL and CL conceived the experiments. YL, DYZ, JBF, ZL, and YH carried out the experiments. YL and DYZ analyzed the data. YL, CL, and XHK wrote the paper. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
The whole study was approved by the Administrative Committee on Animal Welfare of School of Medicine, Nankai University, China (Laboratory Animal Care and Use Committee Authorization, permit number SYXK-2014-0003). All institutional and national guidelines for the care and use of laboratory animals were followed.

 DownLoad:
DownLoad: