













HTML
-
Approximately 71 million people worldwide are infected with hepatitis C virus (HCV), constituting a major global health problem. Approximately 60%–80% of cases progress to chronic infection, which can lead to liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) (World Health Organization Fact sheet, Updated October 2017). HCV infection is treated by a combination of direct-acting antivirals (DAAs) (Heo and Deeks 2018) along with pegylated interferon and ribavirin; although this achieves a high sustained virological response rate, the drugs are unaffordable for many patients (Gottwein et al. 2018; Minichini et al. 2018; Puoti et al. 2018).
HCV replication depends on a variety of host factors (Xu et al. 2012; Guo et al. 2015; Su et al. 2017)—e.g., epigenetic alterations to DNA such as histone methylation and acetylation and the corresponding enzymes—that can be targeted by drugs to attenuate viral replication (Bird 2007; Vaillancourt et al. 2012; Vassilaki et al. 2013). These changes can be induced by aging or pathological conditions such as chronic inflammation or viral infection, leading to inactivation of tumor suppressor genes and tumorigenesis (Shu et al. 2012). Four classes of histone deacetylase (HDAC) with distinct activities have been identified to date, including class Ⅰ (HDAC1, 2, 3, and 8, with strong deacetylase activity) and Ⅱ HDACs; class Ⅲ NAD+-dependent HDACs, also known as sirtuins (SIRTs; SIRT1–7); and HDAC11, the sole member of class Ⅳ (Yang and Gregoire 2005). Class Ⅱ HDACs are further subdivided into Ⅱa (HDAC4, 5, 7, and 9, which cooperate with HDAC1, 2, or 3) and Ⅱb (HDAC6 and 10).
The broad-spectrum HDAC inhibitor suberoylanilide hydroxamic acid (SAHA) and hydroxamic acids can inhibit HCV replication in vitro (Sato et al. 2013; Ai et al. 2015). These inhibitors target multiple HDACs including HDAC1, 2, and 3. However, the activity of specific HDACs in HCV replication is unclear. Since a more targeted drug action is associated with fewer side effects, it is important to distinguish between the functions of different HDAC inhibitors so that the appropriate therapeutic strategy can be applied to a given disease.
Lipid homeostasis is important for the HCV life cycle and is regulated by multiple transcription factors (Bugianesi et al. 2012). Numerous co-factors or -regulators control the expression of genes regulating lipid metabolism. A previous study demonstrated that HDAC3 is an important epigenomic coregulator in hepatic steatosis (Sun et al. 2012), suggesting that it could be a potential drug target for the treatment of HCV infection.
Iron deposition is a feature of chronic HCV infection and contributes to liver injury (Mifuji et al. 2006). Iron withdrawal was shown to improve liver function and suppress HCC development in HCV-infected patients (Kato et al. 2001; Kawamura et al. 2005). Liver-expressed antimicrobial protein (LEAP)-1 (also referred to as hepcidin) is a negative regulator of iron availability that is induced by iron overload and inhibited by iron deficiency in a negative feedback loop (Vecchi et al. 2009). It was reported that HCV inappropriately suppresses LEAP-1 expression, leading to iron accumulation through inactivation of the transcription factors CCAAT/enhancer binding protein α (C/EBPα), hypoxia-inducible factor 1a (HIF1α), and signal transducer and activator of transcription 3 (STAT3) (Miura et al. 2008).
In this study, we investigated the effect of HDAC inhibitor treatment on HCV replication in vitro and in vivo. We studied the mechanism of RGFP966, HDAC3 specific inhibitor, for suppressing HCV replication by regulating Apo-A1 and LEAP-1 expression in Huh7 cells, suggesting that HDAC3 inhibitor could be promising drugs for diseases associated with chronic HCV infection.
-
Huh7 and Huh7-SGR cells (gifts from Prof. Jin Zhong) were cultured in Dulbecco's Modified Eagle's Medium (Gibco, Grand Island, NY, USA) containing 10% heat inactivated fetal bovine serum (Gibco) and 1% penicillin–streptomycin solution (Gibco) at 37 ℃ in humidified air with 5% CO2.
-
HDAC activity was evaluated with the HDAC-Glo Ⅰ/Ⅱ Assay and Screening System (Promega, Madison, WI, USA) according to the manufacturer's protocol.
-
ROS measurement was evaluated with the ROS probe 2', 7'-dichlorodihydrofluorescein diacetate (DCFH-DA) (MedChemExpress, USA) according to the manufacturer's protocol.
-
The HDAC inhibitors SAHA, trichostatin (TSA), CI994I, RGFP966, and PCI-34051 (Selleck Chemicals, Houston, TX, USA) were dissolved in dimethylsulfoxide (DMSO; Sigma-Aldrich, St. Louis, MO, USA) for use in experiments. The inhibitor of HCV NS5B RNA polymerase (2'-C-Methylcytidine, 2'CM, Sigma-Aldrich, St. Louis, MO, USA) was dissolved in ddH2O.
-
Cell proliferation was assessed with CellTiter 96 NonRadioactive Cell Proliferation Assay (Promega) according to the manufacturer's instructions. Briefly, Huh7 cells were seeded in a 96-well plate at a density of 5 × 103/well and cultured for 24 h. The indicated concentrations of RGFP966 were added to the wells followed by incubation for 72 h, and the absorbance at OD490 nm of each well was measured on a microplate reader (Biotek, USA).
-
Total mRNA was extracted from Huh7 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and 100 ng were used for qRT-PCR detection of Apo-A1 and LEAP-1 transcript with SYBR Green PCR Master Mix (Qiagen, Valencia, CA, USA). The primers were listed in Supplementary Table S1. Expression of the micro (mi)RNA miR-122 was evaluated by qRT-PCR using a TaqMan probe; the level was normalized to that of U6 RNA.
-
Total protein was isolated from whole cell lysates and 30–50 mg/lane were separated on a 15% sodium dodecyl sulfate gel and transferred to a nitrocellulose membrane that was probed with antibodies against LEAP-1 (ab30760, Abcam, Cambridge, MA, USA); acetyllysine and growth arrest and DNA damage (GADD)153 (sc-7351, both from Santa Cruz Biotechnology, Santa Cruz, CA, USA); HIF1α, histone H3, acetylhistone H3K9, STAT3, and phosphoSTAT3 (#36169, #4499, #9649, #9139 and #9145 all from Cell Signaling Technology, Danvers, MA, USA); and glyceraldehyde 3-phosphate dehydrogenase (#60004, Proteintech, Chicago, IL, USA). Protein bands were detected by enhanced chemiluminescence.
-
Histone H3 acetylation of Apo-A1, C/EBPα, HIF1α, STAT3, and LEAP-1 was analyzed by ChIP using a commercial kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer's instructions. The primers used are shown in Supplementary Table S1.
-
Procedures involving animals were carried out in accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China) and were approved by the Animal Ethics Committee of Wuhan Institute of Virology. Human CD81 and occludin transgenic (C/OTg) mice (10 to 12 weeks old and matched for age and sex) (Chen et al. 2014) were injected in three batches with 1 mL HCV (JC-1 strain) via the tail vein at a 50% median tissue culture infectious dose of 5 × 107 over 60–120 s to avoid injuring the liver. After 1 week, 2 mg SAHA or RGFP966/kg body weight was administered daily to each animal by intraperitoneal injection for 1–8 weeks. Mice injected with the vehicle DMSO (100 μL/20 g body weight) served as the negative control. Total RNA was extracted from liver or serum using TRIzol (Invitrogen) or TRIzol LS, and qRT-PCR analysis was carried out using the HCV OneStep RT-PCR kit (Qiagen).
-
Data are expressed as mean ± SD of at least three independent experiments and were analyzed using Prism software (GraphPad Inc., La Jolla, CA, USA). Differences between two groups were assessed using unpaired twotailed student t test. For time-series data, the two-way ANOVA procedure was applied. Data involving more than two groups were assessed using ANOVA. P values less than 0.05 were considered significant (*P < 0.05; **P < 0.01 and ***P < 0.001).
Cell Culture
Evaluation of HDAC Activity
Reactive Oxygen Species (ROS) Measurements
Treatment with HDAC Inhibitors
Cell Proliferation Assay
Quantitative Real-Time (qRT)-PCR
Western Blotting
Chromatin Immunoprecipitation (ChIP)
Animal Experiments
Statistical Analysis
-
It was previously suggested that HDAC9 expression was enhanced by HCV infection and that HDAC9 was involved in glucose metabolism, HCV-induced perturbation of glucose homeostasis, and type 2 diabetes (Chen et al. 2015). We examined HDAC expression and activity in HCV- and mock-infected Huh7 cells and found that HDAC9 level was increased 18.02 fold by infection as compared to just 3.28, 3.15, 3.77, and 2.16 fold for SIRT1, HDAC4, HDAC5, and HDAC10, respectively (Supplementary Figure S1). Then we measured the HDAC activity at 72 h after HCV infection. The cytoplasmic and nuclear HDAC activities were enhanced in HCV-infected as compared to mock-infected cells (Fig. 1A); these were abrogated in a dose-dependent manner by treatment with the antioxidants N-acetyl-L-cysteine (NAC) or pyrrolidine dithiocarbamate (PDTC, also an inhibitor of NF-κB) (Fig. 1B). Importantly, suppression of HDAC activity increased histone H3 and histone H3 lysine 9 acetylations in infected cells (Fig. 1C). In addition, HDAC activity was increased in the presence of H2O2, but this effect was abolished by pretreatment with PDTC or NAC (Fig. 1D). ROS production, which was visualized as the fluorescence intensity of the ROS probe DCFH-DA was increased in a time-dependent manner in HCV-infected cells (Fig. 1E). Treatment with TSA or SAHA had no effect on ROS production, suggesting that HDAC acts downstream of ROS. Thus, oxidative stress induced by HCV infection increases HDAC activity, resulting in chromatin remodeling.
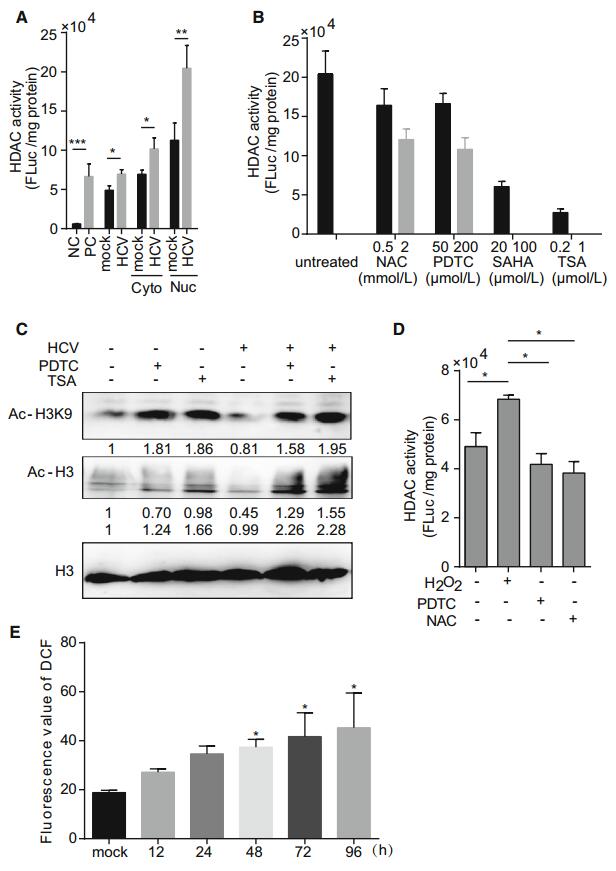
Figure 1. HCV infection increases HDAC activity. (A) HDAC activity in the cytoplasm and nucleus in mock- and HCVinfected Huh7 cells. HeLa cell extract was used as a positive control (PC), and TSA-treated HeLa cell extract was used as a negative control (NC). (B) HDAC activity in HCVinfected Huh7 cells treated with NAC, PDTC, SAHA, or TSA, or left untreated. (C) Western blot analysis of histone H3 (AcH3) and histone H3 lysine 9 (Ac-H3K9) acetylation. Histone H3 was used as an internal control. (D) HDAC activity in mock- and HCV-infected Huh7 cells pretreated with PDTC or NAC for 1 h, then stimulated with H2O2 (200 μmol/L) for 8 h. (E) Fluorescence intensity of 2', 7'-dichlorodihydrofluorescein (DCF) as an indicator of ROS production. Data are presented as mean ± SD of three independent experiments. P values in (A) and (D) were determined by unpaired twotailed student t test and P values in (E) were determined by ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001.
-
We next evaluated whether HDAC inhibitor treatment affects HCV replication by evaluating HCV RNA and protein levels in infected Huh7 cells by qRT-PCR and Western blotting, respectively, following treatment with the various inhibitors for 72 h. SAHA (10 μmol/L, broad-spectrum HDAC inhibitor), CI994I (10 μmol/L, HDAC1/2/3 inhibitor), and RGFP966 (10 μmol/L, HDAC3 inhibitor) suppressed HCV RNA and NS3 expression relative to control cells, whereas PCI-34051 (10 nmol/L, HDAC8 inhibitor) had no effect (Fig. 2A). We investigated the cytotoxicity of the inhibitors in Huh7 cells with the cell proliferation assay and found no change in cell viability following treatment (Fig. 2B). These results suggested that HDACs at least HDAC3 is involved in HCV replication.
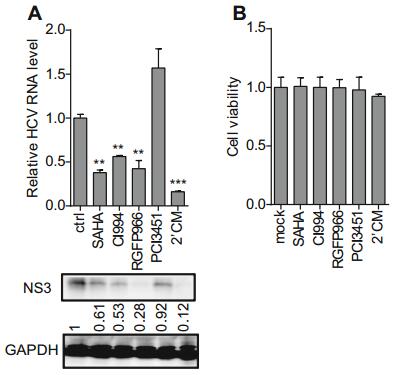
Figure 2. Effect of HDAC inhibitor treatment on HCV replication. (A) HCV replication in Huh7 cells treated with HDAC inhibitors was evaluated by qRT-PCR and Western blotting. DMSO and 2'CM (10 μmol/L, HCV NS5B inhibitor) were used as negative and positive controls, respectively. (B) Viability of Huh7 cells following treatment with HDAC or HCV NS5B inhibitor. Data are presented as mean ± SD of five independent experiments. P values in A were determined by unpaired two-tailed student t test. **P < 0.01; ***P < 0.001.
Although HDAC3 inhibitor treatment suppressed HCV replication, HDAC3 expression in HCV-infected cells was unaffected. To investigate the mechanism by which HDAC3 regulates the HCV life cycle, we evaluated the expression of miR-122, a liver-specific miRNA that was required for HCV replication (Jopling et al. 2005), in Huh7 cells cultured in the presence of 10 μmol/L SAHA, CI994I, or RGFP966 for 72 h. There was no change in miR-122 level upon treatment with these inhibitors, indicating that miR-122 was not involved in the suppressive effect of HDAC3 inhibitor on HCV replication (Supplementary Figure S2).
-
Changes in gene expression by HDAC3 inhibitor treatment were screened by microarray analysis in RGFP966-treated Huh7. The selected genes whose expression level was at least fourfold higher than the level in control cells presented in Supplementary Table S2. Apo-1 and LEAP-1 were selected for further analyzed in this paper. Apo-A1 is involved in the HCV life cycle (Mancone et al. 2011), while LEAP-1, a negative regulator of iron availability, is reduced in HCV patients (Nishina et al. 2008; Lyberopoulou et al. 2015).
The qRT-PCR analysis revealed that the level of LEAP-1 was increased whereas that of Apo-A1 was decreased by treatment with 10 μmol/L RGFP966 for 72 h (Fig. 3A, Fig. 3B). Similar trends were observed in LEAP-1 and ApoA1 protein expression following treatment with 1 mmol/L RGFP966, as determined by Western blotting (Fig. 3C). The effect of RGFP966 on LEAP-1 and Apo-A1 expression was dose dependent (Fig. 3D, 3E).
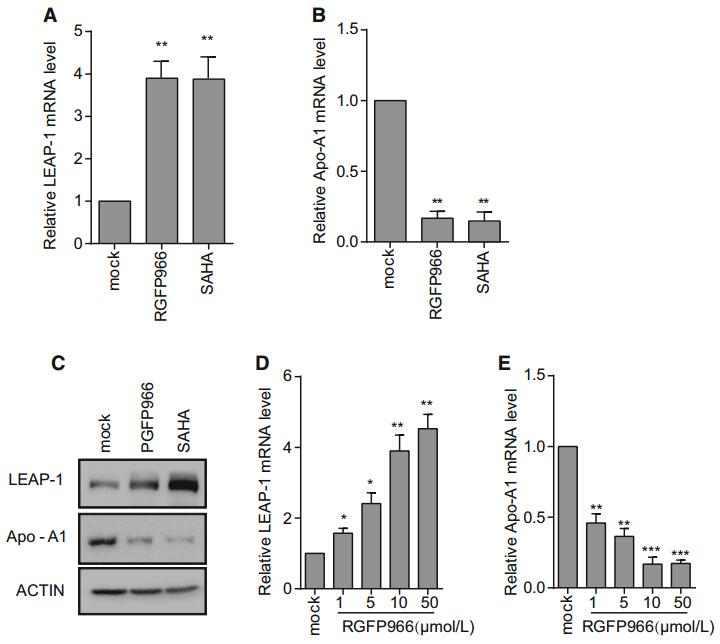
Figure 3. LEAP-1 and Apo-A1 expression in Huh7 cells treated with RGFP966 or SAHA. LEAP-1 and Apo-A1 mRNA and protein levels were evaluated by qRT-PCR (A, B) and Western blotting (C), respectively. (D, E) Cells were cultured in different concentrations of RGFP966 for 16 h, and LEAP-1 and Apo-A1 mRNA levels were evaluated by qRT-PCR and normalized to that of GAPDH. Data are presented as mean ± SD of three independent experiments. P values in (A) and (B) were determined by unpaired two-tailed student t test and P values in (D) and (E) were determined by ANOVA. *P < 0.05; **P < 0.01; ***P < 0.001.
-
The Apo-A1 was significantly up-regulated during HCV infection (Fig. 4A). Histone acetylation typically activated gene expression. We examined whether acetylation of histone binding to LEAP-1 and Apo-A1 promoter was altered in Huh7 cells treated with RGFP966 by ChIP. Since histone H3 acetylation was localized within 500 bp of transcriptional start sites (TSSs), we designed ChIP primers within this region of the LEAP-1 and Apo-A1 promoters. A ChIP-PCR product was obtained only for the Apo-A1 gene that was 78 bp in size. Meanwhile, histone H3 acetylation around the Apo-A1 TSS was increased in RGFP966-treated Huh7 cells (Fig. 4B), although Apo-A1 expression level was decreased.
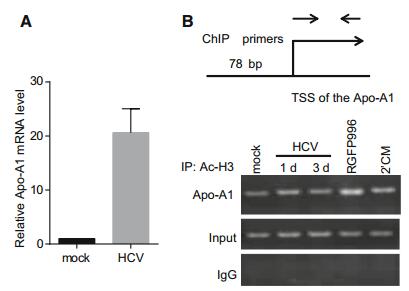
Figure 4. ChIP analysis of Apo-A1 promoter region in Huh7 cells following HDAC3 inhibitor treatment. (A) Apo-A1 mRNA level was evaluated at 72 h after HCV infection by qRT-PCR and normalized to that of actin. (B) Histone H3 acetylation of the Apo-A1 gene promoter after RGFP966 treatment was analyzed using primers located near the TSS, which amplified a 78-bp product.
-
The LEAP-1 promoter region contains the transcription factors C/EBPα, HIF1α, and STAT3 binding sites (Supplementary Figure S3). We first analyzed whether HDAC3 affect C/EBPα bind to the LEAP-1 promoter. C/EBPα binding to the LEAP-1 promoter was reduced in HCVinfected cells, as determined by ChIP (Fig. 5A), while RGFP966 treatment rescued the binding, indicating that HDAC3 activity prevents C/EBPα binding to the LEAP-1 promoter (Fig. 5A). However, the expression levels of C/EBPα and its endogenous inhibitor GADD153 were the same in HCV- and mock-infected cells; also, their expression was unaffected by RGFP966 treatment (Fig. 5B). These results suggest that HDAC3 is involved in the inhibition of C/EBPα binding to the LEAP-1 promoter in HCV infection and the detail mechanism needs to be further revealed.
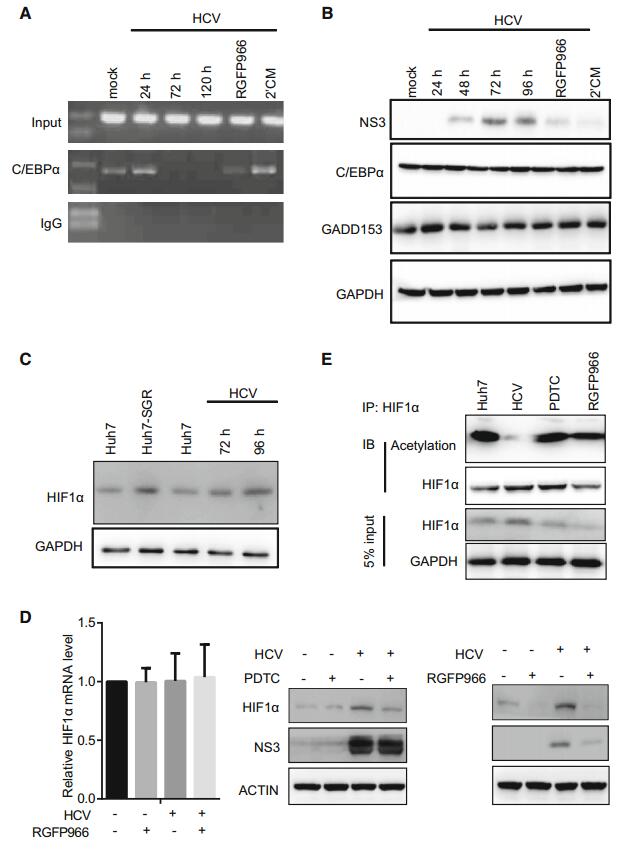
Figure 5. The analysis of C/EBPα and HIF1α binding to the LEAP-1 promoter. (A) ChIP analysis of C/EBPα binding to the LEAP-1 promoter. Input sample and rabbit IgG served as a positive and negative controls, respectively for the PCR reaction. (B) Western blot analysis of C/EBPα and GADD153 expression. GAPDH served as a loading control. (C) HIF1α expression in uninfected Huh7 cells, HCV replicon (Huh7-SGR), or HCVinfected cells, as detected by Western blot analysis (D) HIF1α level was evaluated after incubation in the absence or presence of PDTC (left) and RGFP966 (right) for 16 h by real-time PCR or Western blotting. (E) As in panel (D), total cell lysates were immunoprecipitated (IP) with an anti- HIF1α antibody. Proteins were immunoblotted (IB) with an antibody against HIF1α (lower panel) or acetyllysine (Acetylation; upper panel). Untreated cells served as control (Huh7). Data are representative of three independent experiments.
-
ROS induce HIF1α expression (Schumacker 2005), resulting in the suppression of LEAP-1 expression in mouse liver (Peyssonnaux et al. 2007). HCV infection and replication enhanced HIF1α expression (Fig. 5C); however, RGFP966 treatment of infected cells abolished this effect without affecting HIF1α mRNA levels (Fig. 5D), indicating that a post-translational modification is required for HIF1α degradation, which is known to be induced by acetylation (Ke and Costa 2006). Here, we found that HIF1α acetylation in HCV-infected cells was increased by treatment with RGFP966 (Fig. 5E). These results suggest that HIF1α binding to the LEAP-1 promoter was regulated by HDAC3-mediated deacetylation.
-
STAT3 activation was required for LEAP-1 expression (Pietrangelo et al. 2007). In HCV infection, the levels of cytokines that activate STAT3 were elevated (Zhai et al. 2017), and IL-6 strongly induced LEAP-1 during inflammation. We found here that IL-6 increased LEAP-1 expression 31.22 fold in mock-infected cells, in contrast to a mere twofold increase in HCV-infected cells (Fig. 6A). STAT3 phosphorylation induced by IL-6 in HCV-infected cells was equivalent to that in other groups (Fig. 6B), indicating that this was insufficient for LEAP-1 upregulation. Accordingly, phospho-STAT3 bound weakly to the LEAP-1 promoter in HCV-infected cells, but binding was restored by RGFP966 treatment, as determined by ChIP (Fig. 6C). RGFP966 pretreatment improved the response to IL-6 to a level that was equivalent to that of parent cells (Fig. 6D). STAT3 dimerization, which was important for DNA binding, was regulated by its acetylation (Yuan et al. 2005). As expected, STAT3 acetylation was enhanced by RGFP966 treatment in HCV-infected cells (Fig. 6E).
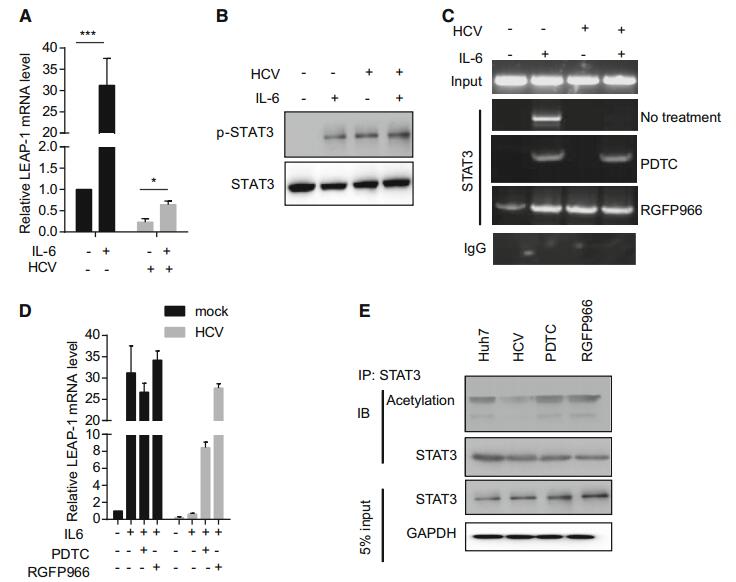
Figure 6. LEAP-1 expression in response to IL-6. Huh7 cells were cultured with 10 ng/mL IL-6 for 16 h. (A) LEAP-1 mRNA level was evaluated by qRT-PCR and normalized to that of actin. (B) STAT3 and phospho-STAT3 protein levels were analyzed by Western blotting. (C) Cells were pretreated with 100 μmol/L PDTC or 10 μmol/L RGFP966 for 8 h, then cultured in the presence of IL-6 for 16 h. ChIP for phospho-STAT3 was carried out, with rabbit IgG serving as a negative control. (D) Cells were pretreated as in panel (C), and LEAP-1 mRNA level was evaluated by qRT-PCR and normalized to that of actin. Data represent mean ± SD. (E) Total cell lysates from HCV-infected cells incubated with 100 μmol/L PDTC or 10 μmol/L RGFP966 for 4 h were immunoprecipitated (IP) with STAT3 antibody. Proteins were immunoblotted (IB) with an antibody against STAT3 (lower panel) or acetyllysine (Acetylation; upper panel). Data are representative of three independent experiments. P values in A were determined by unpaired two-tailed student t test. *P < 0.05; ***P < 0.01.
Together, these results indicate that HDAC3 regulates the binding of C/EBPα, HIF1α, and STAT3 to the LEAP-1 promoter.
-
To confirm the role of HDAC3 in HCV infection in vivo, we evaluated the effect of RGFP966 treatment in immunecompromised humanized transgenic mice harboring both human CD81 and OCLN genes (C/OTg) that were infected with HCV (Chen et al. 2014). To induce persistent infection, mice were intravenously inoculated with HCV JC-1 (genotype 2a) for one week, and then treated daily with SAHA or RGFP966 at 2 mg/kg/day. Both SAHA and RGFP966 suppressed HCV infection in the mice in a time-dependent manner based on serum HCV RNA levels (Fig. 7A, 7C); the HCV genome copy numbers in the liver of treatment mice also were significantly lower than the copy numbers in the control groups in the indicated time point (Fig. 7B, 7D). These results indicated that HDAC3 is involved in HCV infection in vivo.
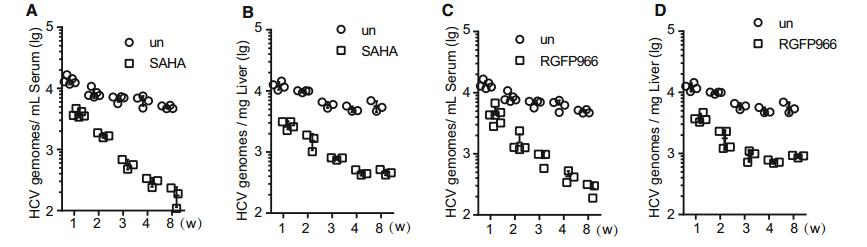
Figure 7. HDAC inhibitor treatment suppresses HCV replication in C/OTg mice. HCV RNA levels in serum (A, C) and liver (B, D) of mice infected with HCV and treated with 2 mg/kg/day SAHA or RGFP966 for 1 to 8 weeks. HCV RNA was detected by qRT-PCR.
ROS Induced by HCV Infection Increase HDAC Activity
HDAC Inhibitor Treatment Suppresses HCV Replication in Infected Huh7 Cells
Apo-A1 and LEAP-1 Expression Is Altered by HDAC3 Inhibitor Treatment
HDAC3 Inhibitor Treatment Increases Histone H3 Acetylation in the Apo-A1 Promoter Region
HDAC3 Prevents C/EBPα Binding to the LEAP-1 Promoter in HCV Infection
HCV-Mediated Stabilization of HIF1α and Downregulation of LEAP-1 Are Abolished By HDAC3 Inhibition
HDAC3 Inhibitor Enhances Interleukin (IL)-6-Induced LEAP-1 Expression via Regulation of STAT3 Acetylation
HDAC Inhibitor Treatment Suppresses HCV Infection In Vivo
-
In this study, we investigated whether HCV infection can be suppressed via epigenetic alterations. Decreasing acetylation by treatment with HDAC3 inhibitor suppressed ApoA1 and stimulated LEAP-1 expression in HCV-infected cells. Depressed levels of Apo-A1—a protein component of high-density lipoprotein particles that was thought to be necessary for HCV particle formation and maintenance of infectivity—will suppress HCV assembly and secretion. On the other hand, increased LEAP-1 levels may remit HCV infection and chronic liver injury. HDAC inhibitors have been shown to block HCC progression (Lu et al. 2007); we found here that HCV replication was diminished by HDAC inhibitor treatment.
HCV-induced ROS is one of the factors to activate HDACs with subsequent acetylation of histones. It has been reported weak oxidative stress induces hyperacetylation, whereas strong oxidative stress to induce deacetylation even (Berthiaume et al. 2006). Therefore, the relationship between ROS, HDAC activity, and acetylation status may depend on the intensity of oxidative stress, or endogenous HDAC activity. Meanwhile, we noticed although anti-oxidants markedly reduced ROS production in HCV infection, its effect in HDAC inactivation was less compared with a HDAC inhibitor, suggesting that HDAC is activated by other factors in addition to ROS. Whether HCV regulates HDAC activators, including protein kinase CK2 and 14-3-3 proteins which are important for the nuclear localization of class Ⅱ HDACs, is potential causes (Aoki et al. 2000; Sengupta and Seto 2004; Ivanov et al. 2011).
HIF1α deacetylation leads to ubiquitination-dependent protein degradation (Ke and Costa 2006). HIF1α loaded by HDAC3 translocates to the nucleus where it becomes functionally activated (Tovar-Castillo et al. 2007). HDAC3 inhibitors can prevent cancer development by inactivating HIF1α target genes. We demonstrated that HDAC3 regulates HIF1α stability in HCV-infected cells as well as the acetylation of HIF1α, thereby altering their ability to bind to the LEAP-1 promoter. HDAC3 also prevented the binding of C/EBPα to the LEAP-1 promoter, resulting in decreased LEAP-1 transcription. Other studies have reported that C/EBPα promoter binding is attributable to its acetylation (Lu et al. 2007). The level of IL-6, which stimulates LEAP-1 expression, was elevated in the serum of HCV-infected patients and mice. HCV-infected cells showed impaired LEAP-1 expression in response to IL-6 despite STAT3 phosphorylation; however, this was reversed by HDAC3 inhibitor pretreatment, indicating that C/EBPα and STAT3 binding to the LEAP-1 promoter involves modulation of acetylation status by HDAC3.
Our experiments in mice showed that SAHA or RGFP966 treatment was effective in suppressing HCV infection and was well tolerated. RGFP966 specifically targets HDAC3 and therefore has fewer side effects than SAHA, which also inhibits HDAC1 and 2. This is the first study demonstrating that HDAC3 inhibitor treatment can suppress HCV replication, an effect that was likely mediated via downregulation of Apo-A1 and upregulation of LEAP-1. Our findings suggested that epigenetic therapy with an HDAC3 inhibitor such as RGFP966 can be an effective strategy for treating diseases associated with HCV infection such as HCC.
-
We would like to thank the members of Chen's lab and the core facilities center of Wuhan institute of virology for technical help. This study was supported by the National Key Research and Development Program of China to YZ (2018YFA0507202) and the Program for Youth Innovation Promotion Association in Chinese Academy of Science to JC.
-
JC conceived/designed the experiments. YZ and QW performed the experiments and analyzed the data. YQ, JT, XC and DG contributed reagents/materials/analysis tools. JC and XC wrote the manuscript. YZ and QW prepared the figures and tables. JC checked and finalized the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Author Contributions
-
The authors declare that they have no conflict of interest.
-
The study was approved by the Animal Ethics Committee of Wuhan Institute of Virology. All institutional and national guidelines for the care and use of laboratory animals were followed.
Conflict of interest
Animal and Human Rights Statement
-

Table Supplementary Table S1. The primers in RT-PCR and CHIP

Table Supplementary Table S2. Changes in gene expression by HDAC3 inhibitor treatment
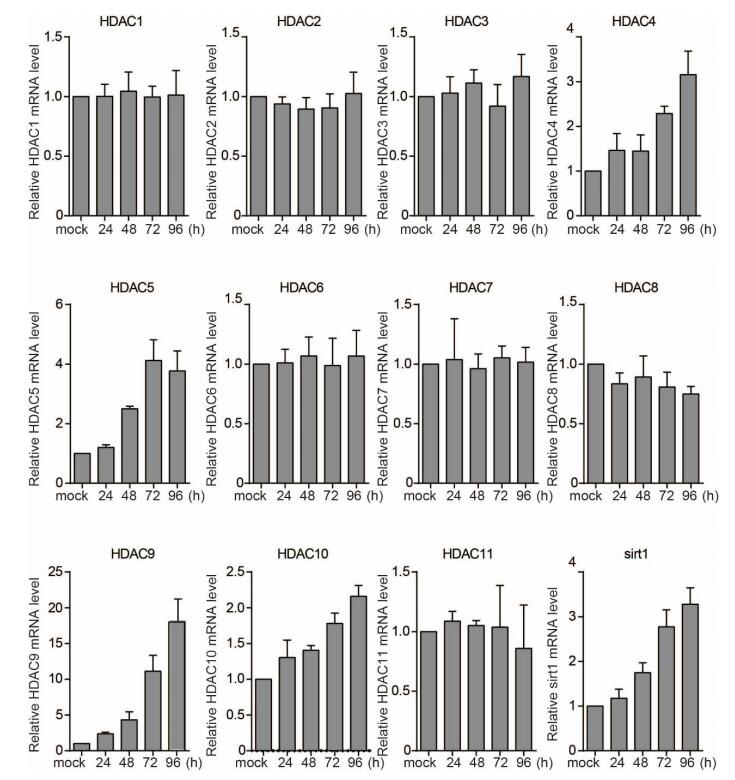
Figure SupplementaryFigureS1. HDAC1, HDAC2, HDAC3, HDAC4, HDAC5, HDAC7, HDAC9, HDAC11, and SIRT1 mRNA levels in mock- or HCV-infected-HuH7 cells at indicated time points post infection
Figure SupplementaryFigureS2. miR-122 expression in Huh7 cells following treatment with SAHA, CI994I, or RGFP966. Huh7 cells were treated with 10 μmol/L SAHA, CI994I, or RGFP966 for 72 h, and miR‐122 levels normalized to that of U6 were evaluated by qRT‐PCR. Data represent mean ± SD of three experiments.
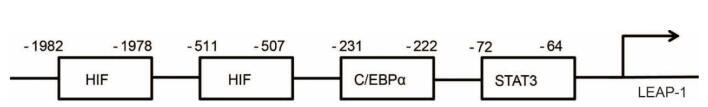
Figure S3. Human LEAP-1 promoter sequence with putative transcription factor binding sites.

 DownLoad:
DownLoad: