














HTML
-
Influenza A viruses of the H5 subtype can infect multiple species of poultry, wild birds, and mammals. In recent years, H5 subtype viruses have been continuously circulating worldwide. The spread of H5 highly pathogenic avian influenza (HPAI) viruses is a serious threat to the poultry industry, wild bird ecology and human health. H5 subtype viruses have high genetic diversity and a very high rate of evolution (Duan et al. 2008; Vijaykrishna et al. 2008; Guan et al. 2009). Based on the sequence of the hemagglutinin (HA) gene, H5 subtype viruses can be divided into different clades (WHO et al. 2008). Viruses in different clades have different geographical distributions.
In China, most clade 2.3.2 viruses clustered in Hong Kong, Henan, Inner Mongolia and Qinghai (Smith et al. 2009; Li et al. 2011; Bi et al. 2015; Bi et al. 2016a), while clade 2.2 viruses have persisted for long periods in Qinghai Lake (Chen et al. 2005). Clade 2.3.4 viruses first emerged in the south and subsequently spread to the north (Tian et al. 2015a). In contrast, clade 7 viruses were first isolated in northern China and spread from north to south into Hunan and Yunnan provinces (Tian et al. 2015a). A study of the spatiotemporal patterns of H5N1 HPAI viruses in China found a significant time lag between poultry or wild bird outbreaks and human cases. Viruses circulating in poultry spread faster, but were more spatially clustered (Tian et al. 2015b). The spread of H5 viruses in wild birds is associated with bird migration networks and the peak of migration (Tian et al. 2015b).
In 2014, a new clade of H5 viruses (2.3.4.4) emerged. This clade includes H5N1, H5N2, H5N3, H5N5, H5N6 and H5N8 subtype viruses (WHO 2015). In 2014, a novel H5N6 virus was first reported in China (Pan et al. 2015). To date, a total of 18 countries have reported cases of H5N6 infection, including China, Korea, Japan, Vietnam, Laos, Myanmar, the Philippines, Greece, Sweden, Switzerland, Germany, Finland, Denmark, Iran, Ireland, Netherlands, Slovakia and the United Kingdom (OIE 2019). H5N6 viruses have become the predominant avian influenza virus subtype circulating in southern China. This virus was derived from reassortment between the internal genes of H9N2/H7N9 viruses and a clade 2.3.2.1c H5N1 virus (Bi et al. 2016b; Yang et al. 2017). In 2014, a novel H5N8 virus caused outbreaks in domestic ducks and wild birds in South Korea (Lee et al. 2014). As these birds migrated, the H5N8 virus spread to Japan, Siberia, North America and Europe (Lee et al. 2015; The Global Consortium for H5N8 and Related Influenza Viruses 2016). In 2016, a novel reassortant H5N8 virus bearing internal genes from a low pathogenic avian influenza virus circulating in Mongolia was identified in wild birds in Qinghai Lake (Li et al. 2017). In North America, novel reassortant H5N1, H5N2 and H5N8 viruses bearing genes of local low pathogenic avian influenza viruses were identified (Lee et al. 2016). In Chinese Taiwan, novel reassortant H5N2, H5N3 and H5N8 viruses bearing Eurasian influenza genes were identified (Huang et al. 2016). In Chinese mainland, novel reassortant H5N5 viruses bearing internal genes from H5N1 viruses were identified (Gu et al. 2011).The genes of HPAI H5 clade 2.3.4.4 viruses around the world show significant diversity and bear internal genes from multiple viral subtypes (El-Shesheny et al. 2017; Kim et al. 2017).
The evolutionary patterns of HPAI H5 viruses in China are not well understood. Studies in this area would help us to understand the geographical dispersal of H5 viruses and to identify areas that are significant for viral migration and evolution.
-
Full-length HA sequences of H5 HPAI viruses in China were obtained from the GISAID (http://gisaid.org) and the GenBank (http://www.ncbi.nlm.nih.gov/genomes/FLU/) repositories on 1 January 2019. HA sequences were aligned and neighbor-joining (NJ) phylogenies were estimated using MEGA 7 software (https://www.megasoftware.net/). NJ methods were used to perform system reconstruction and to calculate confidence values to estimate the number of repetitions (1000). Residual analyses of phylogenetic trees were performed using TempEst v1.4 (http://tree.bio.ed.ac.uk/software/tempest/), and anomalous sequences were removed based on the estimated root-to-tip distance. To control sampling biases, we removed sequences with nucleotide similarities greater than 99% within the same year and region to ensure fewer than 50 sequences per year were used. After removing duplicate sequences, we analyzed 398 HA gene sequences from poultry (Supplementary Table S1) and 133 HA gene sequences from wild birds (Supplementary Table S2) isolated between 2000 and 2018. All sequences were used to analyze the dispersal of H5 HPAI viruses in China. We collected data on HPAI H5 virus outbreaks in China from the EMPRES Global Animal Disease Information System (Claes et al. 2014). The outbreak data included information on time, latitude and longitude, and infected hosts.
-
The study area included twenty-seven provinces, municipalities and autonomous regions in China. The HA sequences from wild birds were distributed across thirteen regions including the Hong Kong Special Administrative Region of China and the provinces of Qinghai, Henan, Heilongjiang, Inner Mongolia, Jilin, Jiangxi, Shanghai, Hubei, Jiangsu, Guangdong, Guangxi and Hunan. The HA sequences from poultry were distributed across twenty-six regions including the Hong Kong Special Administrative Region of China and the provinces of Zhejiang, Yunnan, Xinjiang, Tibet, Qinghai, Sichuan, Shanxi, Shanghai, Shandong, Ningxia, Liaoning, Jilin, Jiangxi, Jiangsu, Hunan, Hubei, Henan, Hebei, Guizhou, Guangxi, Guangdong, Gansu, Fujian, Anhui and Heilongjiang.
-
A continuous time Markov chain Monte Carlo (MCMC) model was constructed to estimate time-resolved phylogenetic trees using BEAST 1.8.2 software (Drummond et al. 2012). For all runs, the SRD06 nucleotide substitution model (Shapiro et al. 2006) was used to estimate the divergence times of viruses, along with the uncorrelated log-normal relaxed clock model and a Gaussian Markov random field (GMRF) Bayesian Skyride coalescent prior (Minin et al. 2008). The MCMC chain was run for 500 million iterations, with a 10% burn-in and sub-sampling every 50, 000 iterations. GMRF Bayesian Skyride analysis was used to elucidate the population dynamics of H5 viruses.
The discrete-state continuous time Markov chain method was used to estimate the transition rates of H5 viruses between discrete sampling locations. A Bayesian stochastic search variable selection model with asymmetric substitution was used. The effective sample size values of the pairwise migration rate estimates were > 200 using Tracer v1.6 (Rambaut et al. 2018). We visualized and annotated phylogenetic trees in Figtree (http://tree.bio.ed.ac.uk/software/figtree/) and visualized geographic transition routes in SPREA D3 (version 0.9.7.1) (Bielejec et al. 2016). The Bayes factor (BF) provided statistical support for geographic transition routes. BF values > 100 indicated robust statistical support. BF values > 30 and ≤ 100 indicated very strong statistical support. BF values > 10 and ≤ 30 indicated strong statistical support. BF values > 6 and ≤ 10 indicated substantial statistical support. BF values > 3 and ≤ 6 indicated poor statistical support (Lemey et al. 2009).
Virus Sequence Data
Study Area
Phylogenetic Analysis
-
We constructed a Bayesian genealogical tree to display the time scale of H5 virus HA gene evolution in poultry in China. Twenty-six geographical regions were assigned different colors in the phylogenetic tree (Fig. 1). H5 viruses originated in various provinces in China, and showed complex cladistics and rapid evolution. The genealogical tree showed that H5 viruses isolated from poultry belonged to clades 0, 1, 3, 4, 5, 6, 7, 7.2, 8, 9, 2.4, 2.5, 2.3.1, 2.3.2, 2.3.2.1, 2.3.4 and 2.3.4.4. H5 HPAI viruses were first isolated in Guangdong Province, and then rapidly spread northward, eastward and westward. These viruses evolved quickly, with clade 2.3.2 giving rise to clades 2.3.2.1a, b and c, clade 7 giving rise to clade 7.2, and clade 2.3.4 giving to clades 2.3.4.4.a, b, c and d.

Figure 1. Phylogenetic tree of H5Nx HA sequences isolated from poultry. The tree is colored based on the twenty-six geographical regions in which the viruses were sampled. The black points represent nodes, and the horizontal bar indicates the 95% highest posterior density of each node.
We also constructed a Bayesian genealogical tree to display time scale of H5 virus HA gene evolution in wild birds in China. Thirteen geographical regions were assigned different colors in the phylogenetic tree (Fig. 2). H5 viruses originated in various provinces in China, and again showed complex cladistics and rapid evolution. The genealogical tree showed that H5 viruses isolated from wild birds belonged to clades 1, 3, 4, 6, 7, 9, 2.2, 2.3.2, 2.3.2.1, 2.3.4 and 2.3.4.4. H5 HPAI viruses were first isolated in Guangdong Province in 2002, and subsequently spread rapidly northward and westward. Clade 2.2 viruses were first isolated in Qinghai Province and were not found in other provinces in 2005. The viruses evolved quickly, with clade 2.3.2 giving rise to clades 2.3.2.1b and c and clade 2.3.4 giving rise to clades 2.3.4.4.a, b, c and d.
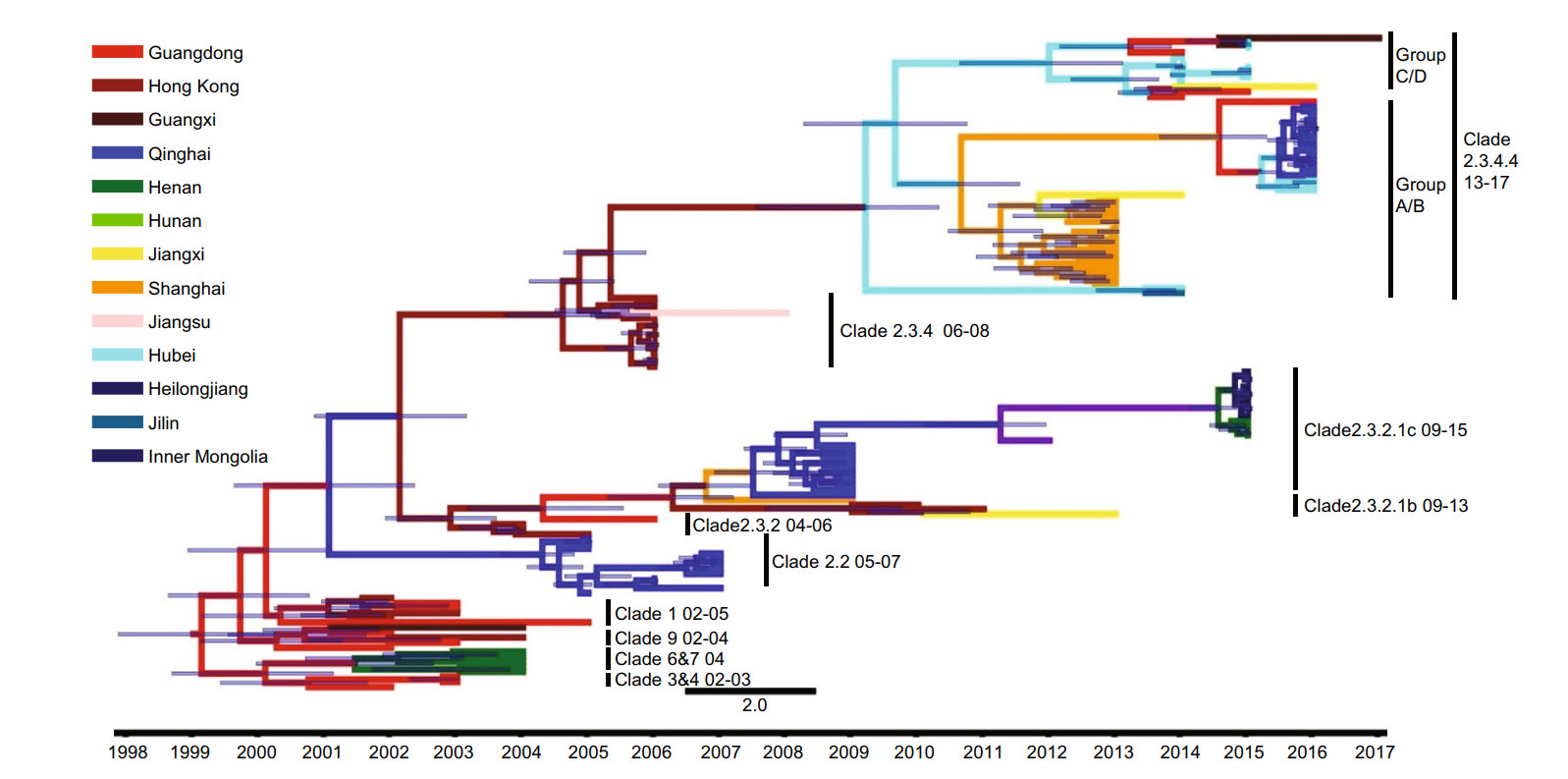
Figure 2. Phylogenetic tree of H5Nx HA sequences isolated from wild birds. The tree is colored based on the thirteen geographical regions in which the viruses were sampled. The black points represent nodes, and the horizontal bar indicates the 95% highest posterior density of each node.
-
We reconstructed the spatial dispersal networks of HPAI H5 viruses in poultry and wild birds in China. Overall, we found that the transmission routes of viruses in poultry and wild birds were quite different. Transmission routes with BF values > 10 were selected for analysis. There were 46 significant transmission routes of viruses in poultry (Supplementary Table S3). Guangdong, Hunan, Shanghai and Jiangsu acted as the epicenters for the spread of various H5 subtypes viruses in poultry (Fig. 3). Guangdong was linked with ten locations (Fujian, Guangxi, Hong Kong, Hubei, Hunan, Anhui, Jiangsu, Liaoning, Shanxi, and Heilongjiang), Jiangsu was linked with eleven locations (Guangdong, Guangxi, Jiangxi, Shanghai, Tibet, Hong Kong, Hubei, Hunan, Jilin, Heilongjiang, and Zhejiang), Shanghai was linked with eight locations (Hebei, Hunan, Sichuan, Heilongjiang, Jiangsu, Jiangxi, Ningxia, and Shandong), and Hunan was linked with seven locations (Guangdong, Jilin, Shanghai, Heilongjiang, Jiangsu, Jiangxi, and Anhui). There were 14 major transmission routes of viruses in wild birds (Supplementary Table S4). Henan Province, Shanghai, Hong Kong and Inner Mongolia acted as epicenters for the spread of various H5 subtypes viruses in wild birds (Fig. 4). Henan was linked with three locations (Hunan, Shanghai, and Inner Mongolia), Shanghai was linked with three locations (Jilin, Jiangxi, and Henan), Hong Kong was linked with three locations (Jilin, Jiangsu, and Inner Mongolia), and Inner Mongolia was linked with four locations (Henan, Heilongjiang, Hong Kong, and Jiangsu).
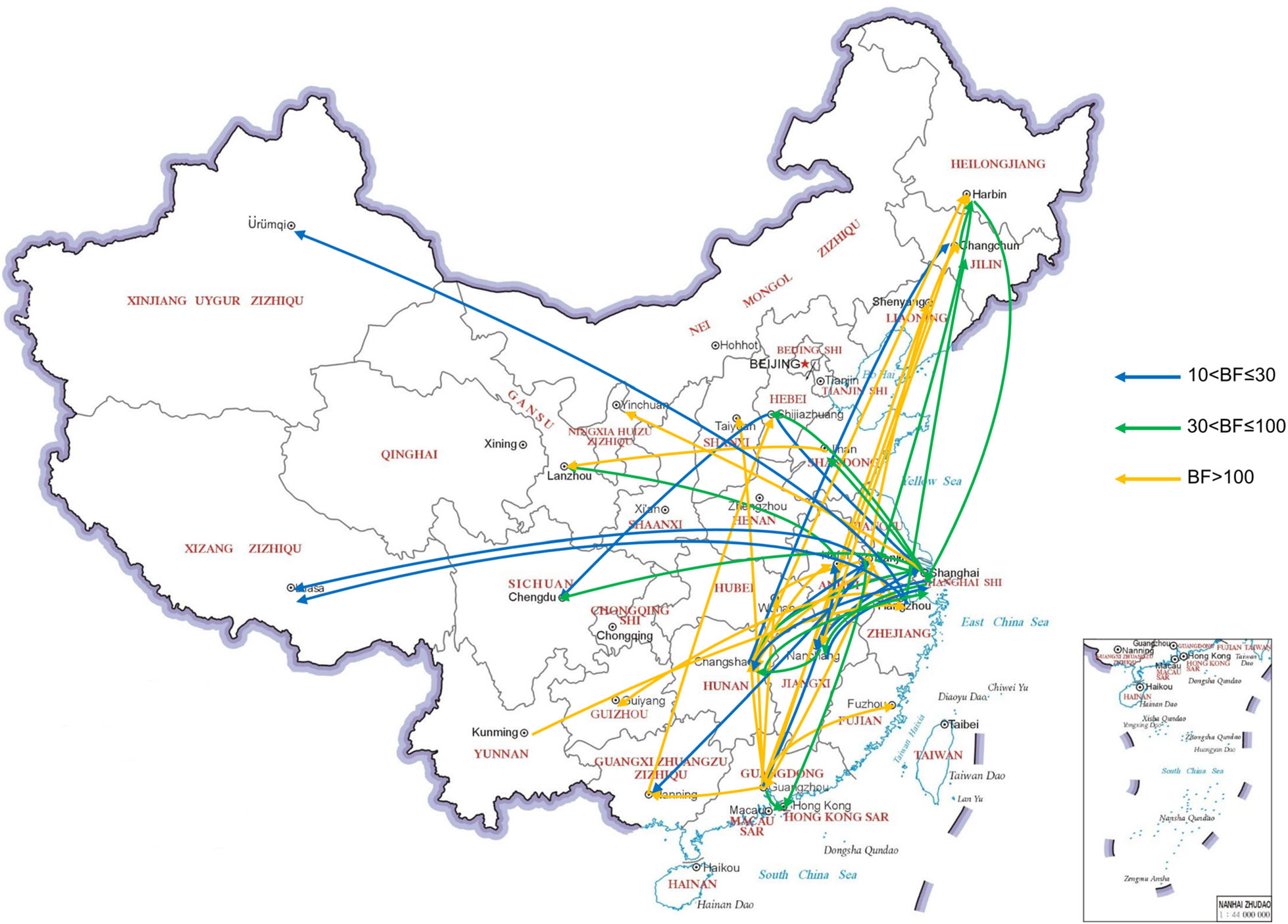
Figure 3. Migration routes of H5Nx viruses isolated from poultry with in twenty-six geographic regions in China. The red, green and blue arrows indicate the reconstructed spatial dispersal patterns of H5Nx viruses isolated from poultry in China. The Bayes factor (BF) test provides statistical support for geographic transition routes. BF values > 100 indicate robust statistical support. BF values > 30 and ≤ 100 indicate very strong statistical support. BF values > 10 and ≤ 30 indicate strong statistical support.

Figure 4. Migration routes of H5Nx viruses isolated in wild birds within thirteen geographic regions in China. The red, green and blue arrows indicate the reconstructed spatial dispersal patterns of H5Nx viruses isolated from wild birds in China. The Bayes factor (BF) test provides statistical support for geographic transition routes. BF values > 100 indicate robust statistical support. BF values > 30 and ≤ 100 indicate very strong statistical support. BF values > 10 and l ≤ 30 indicate strong statistical support.
-
We inferred H5 virus demographic history using the GMRF Bayesian Skyride model to examine changes in genetic diversity from 2000 to 2018 in China. In poultry, the effective population size fluctuated slightly from 2000 to 2012, and from 2012 to 2014, genetic diversity rapidly increased. After 2014, the effective population size decreased year by year (Fig. 5). In wild birds, the effective population size decreased gradually from 2000 to 2005, and from 2005 to 2013, the effective population size increased slowly. From 2013 to 2015, genetic diversity decreased, and after 2015, the effective population size remained constant (Fig. 6).
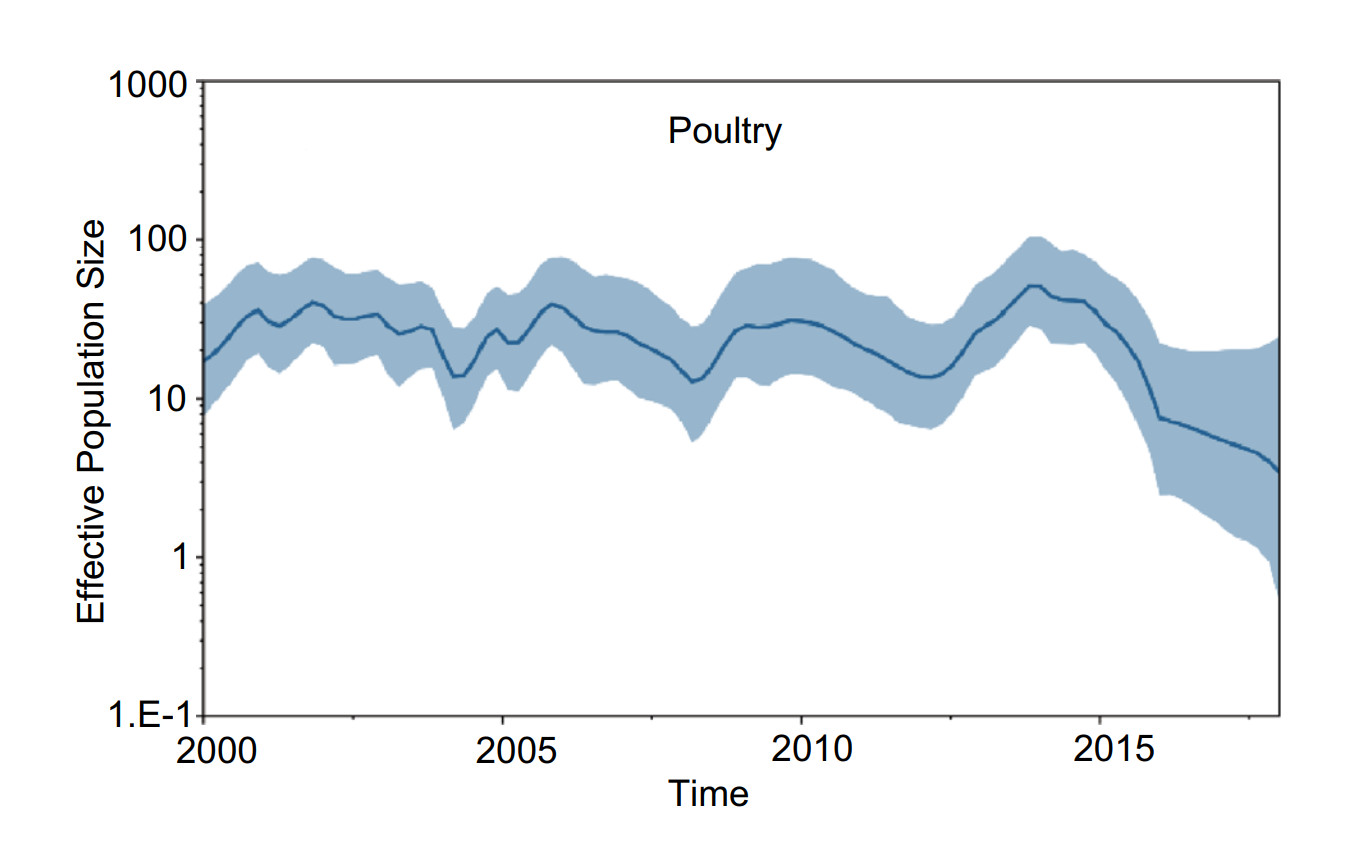
Figure 5. Gaussian Markov random field (GMRF) Bayesian Skyride plot of H5Nx viruses isolated in poultry in China. The plot was constructed from HA. A GMRF Bayesian Skyride analyses of HA genes to display changes in the effective population size over time. The solid blue line indicates the median value, and the shaded blue area represents the 95% highest posterior density of genetic diversity estimates.

Figure 6. Gaussian Markov random field (GMRF) Bayesian Skyride plot of H5Nx viruses isolated in wild birds in China. The plot was constructed from HA. A GMRF Bayesian Skyride analyses of HA genes to display changes in the effective population size over time. The solid blue line indicates the median value, and the shaded blue area represents the 95% highest posterior density of genetic diversity estimates.
Phylogenetic Analysis of H5 Viruses in China
Spatiotemporal Dynamics of H5 Virus Geographic Dispersal
Population Dynamics during the Geographical Dispersal of H5 Viruses
-
In this study, we investigated the transmission and evolution of HPAI H5 viruses in poultry and wild birds in China. We found that H5 HPAI viruses exhibited distinct evolutionary dynamics in poultry and wild birds. Bayesian phylogeographic analysis of H5 viruses showed that Guangdong Province was the origin for the spread and migration network of these viruses in poultry and wild birds. Guangdong, Jiangsu, Shanghai and Hunan acted as epicenters for the spread of various H5 subtypes viruses in poultry, and Henan, Shanghai, Hong Kong and Inner Mongolia acted as epicenters for the spread of various H5 subtypes viruses in wild birds.
HPAI H5 viruses have circulated widely in Asian countries over the past decade (Chen et al. 2006; Smith et al. 2006). China is one of the countries seriously threatened by HPAI H5 viruses (Wu et al. 2014). The viral transmission network in poultry showed long-distance migration links in the north–south and east–west directions as well as short-distance migration links between adjacent geographical regions. Studies have shown the HPAI viruses spread over long distances through live poultry movement and spreads short distances through the poultry market (Li et al. 2018). Guangdong, Jiangsu, Shanghai and Hunan acted as epicenters for the spread of various H5 subtypes viruses in poultry. Poultry trade and transportation are well developed in Guangdong, Jiangsu, Shanghai and Hunan. Backyard poultry farming in southern China is high-density and free-range. Infected poultry in backyard farms could spread the virus through transportation (Van Kerkhove et al. 2009) and live poultry trading (Kilpatrick et al. 2006). Live poultry markets are a high-risk areas for transmission and spread of HPAI viruses (Lam et al. 2013). Many birds are placed in close proximity in these markets, increasing the probability of emergence of novel viruses and of poultry–poultry and poultry–human transmission (Leung et al. 2012; Su et al. 2015). Live poultry markets and backyard farms lack biosafety measures. These highrisk environmental factors can cause the spread of HPAI viruses. Increased biosafety measures in the poultry industry and surveillance of H5 HPAI viruses in these four regions could reduce the risk of a future epidemic.
Henan, Shanghai, Hong Kong and Inner Mongolia acted as epicenters for the spread of various H5 subtypes viruses in wild birds. Studies have shown that Henan and Inner Mongolia are high-risk areas for viral transmission in wild birds along the flyways (Bi et al. 2015, 2016a). The main migration route of HPAI H5 viruses in Chinese migratory birds are from south to north and from north to south. This may be due to long-distance migration of migratory birds in the East Asia-Australia flyway as well as short-term foraging activities. The East Asia-Australia migration zone is the most significant migration route passing through China. Each year, 50 million waterfowl, including more than 55 migratory species, migrate through this zone (Olsen et al. 2006; Flyway 2011; Flyway 2015). Avian influenza viruses are host-specific and these viruses can persist in aquatic environments. Avian influenza viruses can be transmitted among poultry and wild birds living in the same aquatic environments through the fecal–oral route (Bertran et al. 2014). Wild birds acquire HPAI H5 viruses from poultry and transport the viruses to more distant locations. Studies have shown that migratory birds first gather in the vicinity of the 0 ℃ isotherm (Reperant et al. 2010), and then carry out migratory activities, leading to the geographical spread of the virus. During geographical spread, the virus is constantly evolving to accommodate the long-distance migratory activities of birds. In China, the spread of HPAI H5 viruses in wild birds may be caused by migration of wild birds.
This study had several limitations. First, sampling bias may have affected the Bayesian phylogenetic reconstruction and inference of transmission networks. Second, because gene sequence data and the road length data for each province were not matched by time, we could not compare our results with poultry transportation data. Third, the spatiotemporal information for HA sequences used in the article may not have accurately represented the times and locations in which wild birds were infected with H5 HPAI viruses. Some wild birds are carriers of these viruses in the absence of clinical symptoms (Brown et al. 2008; Keawcharoen et al. 2008). This could result in increased viral spread by migratory birds on a large scale, a possibility that was not considered in our study.
-
This study was partially supported by the National Research Program of the Ministry of Science and Technology of the People's Republic of China (2016YFA0600104) and by donations from Delos Living LLC and the Cyrus Tang Foundation to Tsinghua University.
-
XWL and BX conceived and designed the research. XYL provided helpful suggestions regarding the study. XWL and BX wrote the manuscript.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: