











-
Duck hepatitis virus (DHV) causes a highly contagious disease in young ducks which often associated with liver necrosis, hemorrhages and high mortality. The disease was first described in Long Island in 1949 (7). Subsequently, outbreaks were reported from England, Canada, Germany, Japan and elsewhere. In 1963, the disease emerged in Mainland China, although its pathogen was not identified until 1984. Three distinct serotypes of DHV (DHV-1 to 3) have been described and classified as picornaviruses (4, 11). No antigenic relationships exist among the 3 serotypes (8, 14), of which DHV-1 is distributed worldwide and thus has the most economic relevance. DHV-1 was originally classified as an enterovirus (13). Later on, it was found to be more closely related to the genus Parechovirus than to other picornaviruses (6), therefore, it was suggested that it be classified as a new genus within the Picornaviridae family (12).
The complete genome of DHV-1 has been determined for the Korea (6) and Taiwan isolates (12), which comprise a single-stranded positive-sense RNA genome of 7 691 nucleotides (nt) with a poly(A) tail at the 3′-end. The genomic organization of DHV-1 is similar to other picornaviruses; there is only one large open reading frame (ORF) in the whole genome, which is flanked by 5′-and 3′-non-coding regions (UTRs). But the genome of DHV-1 shared low sequence identity with other picornavi-ruses and the phylogenetic analyses revealed that DHV-1 was a distinct picornavirus species.
To date, only one attenuated DHV-1 Chinese isolates has been reported (1), however, the sequence of virulent DHV-1 from mainland of China remains unknown. In the present paper, we reported the complete sequence of a virulent DHV-1 ZJ-V isolate isolated from Zhejiang Province, and analyzed its genetic relationship with those reported by Tseng et al (12).
HTML
-
Baby hamster kidney cell line (BHK-21) was maintained in Eagle's Minimal Medium (EMEM) supplemented with 10% fetal bovine serum (FBS). The original DHV-1 isolate (ZJ-V) was isolated from a duck farm in Zhejiang Province, China. After passaging 20 times in chicken embryos, ZJ-V was propagated on BHK-21 cells in EMEM with 2% FBS. After 72 h of incubation at 37℃, when more than 80% of the cells showed cytopathogenic effects, the cells were subjected to 3 cycles of freeze-thaw. The viral supernatant was clarified by centrifugation at 800 × g for 10 min and stored at −80℃.
-
Genomic RNA was extracted from the viral supernatant with RNAeasy (Qiagen, Germany), and used immediately for cDNA synthesis. cDNA synthe-sis was performed with SuperScript Ⅱ reverse trans-criptase (RT) (Invitrogen, USA) and specific RT primers (Table 1). A total of 5 fragments, covering the complete genome of DHV-1, were subsequently PCR amplified with Pfu Turbo DNA polymerase according to the manufacturer's protocol (Stratagene, USA).

Table 1. Sequences of the primers used for amplification of a full-length genome of ZJ-V a
The cDNA fragment from the 3′-end of the viral genome was amplified by the Rapid Amplification of cDNA Ends (RACE) method. The RACE kit was purchased from TaKaRa(Japan)and the procedure was carried out according to the manufacturer's protocol. The remaining sequence of ZJ-V was amplified using specific primers at several positions along the tem-plate RNA based on the published DHV-1 sequence.
-
Each purified PCR product was sequenced 3 times either directly or after ligation to the pGEM-T Easy vector (Promega, USA). Cycle sequencing reactions were performed using the Thermo Sequenase Cycle Sequencing Kit (Amersham Pharmacia Biotech, Sweden) or the Thermo Sequenase Cy5.5 Dye Terminator Cycle Sequencing Kit (Amersham Phar-macia Biotech, Sweden) according to the manu-facturer's instructions.
-
DHV-1 RNA structure was analyzed with mfold, version 3.1, under default folding conditions (37℃, 1 mol/L NaCl, and no limit on distance between paired bases). Secondary-structure plots were generated with RnaViz, version 2.0.
-
The sequences of nucleotide and deduced amino acids were compared and analyzed using DNAstar software (Lasergene). Multiple alignments between ZJ-V and other isolates genes and proteins were performed with and analyzed by DNAStar MegAlign program (version 4.0.3) with the PAM250 amino acid substitution matrix and Joltun Hein method and the following parameters: gap opening penalty = 11 and gap extension penalty = 3. Identity values were determined from the generated alignments.
A phylogenetic tree was constructed by the MegA-lign program of DNAstar software on the basis of amino acid sequence alignment of DHV-1 VP1.
-
The sequences of VP1 of ZJ-V and other isolates were compared. These reference isolates were DRL-62 (DQ219396), O3D (DQ249299), H (DQ249230), 5586 (DQ249301), R85952 (DQ226541), C80 (EF093502), JX (DQ864514), ZJ (EF382778), SY1 (EF407857), SY2 (EF407858), SY3 (EF407859), SY4 (EF407860), SY5 (EF407861), SY6 (EF407862), SH-A (EF502171), ZJ-A (EF502172), ZJ-3 (EF-502170), ZJ07-2 (EF502169), ZJ07-1 (EF502168), AV2111 (EF442073) and ZJ-A2 (EF442072).
Cells and virus isolate
RNA extraction and RT-PCR
Sequencing
RNA structure prediction.
Comparative sequence analysis
GenBank accession numbers
-
The full-length genome of ZJ-V isolate was sequenced with 5 pairs of specific primers (Table 1), and the results showed that the complete nucleotide sequence of ZJ-V consisted of 7 691 nt (excluding the poly (A) tail) and contained a single ORF of 6 747 nt terminating in a UGA codon. The 3′-terminal sequence of ZJ-V isolate was amplified by the 3′-RACE, and the results showed that the poly (A) tail of DHV I had at least 22 As. The probable genomic organization of ZJ-V is shown in Fig. 1. Sequence alignment revealed that the DHV-1 ZJ-V isolate polyprotein contained a 3Cpro protease and potential proteolytic cleavage sites. The final results of processing suggested that approxi-mately 12 proteins (VP0/VP3/VP1/2A1/2A2/2A3/2B/ 2C/3A/3B/3C/3D) were released. These included 3 copies of the 2A gene, with a NPG↓P cleavage site between 2A1 and 2A2 (Fig. 1), similar to that in the genus Aphthovirus (16). The genomic sequence of ZJ-V has been submitted to GenBank and its accession number is EF382778.
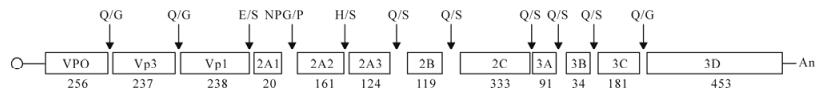
Figure 1. Genome organization and diagrammatic representation of the primary cleavage products released by 3A protease activity during polyprotein processing of the DHV-1 polyprotein. The potential cleavage sites for DHV-1 are shown as dipeptides above the protein products with the length of amino acids (aa) below (not to scale).
-
No nucleotide insertions or deletions were observed in the genomic sequence of ZJ-V when compared with that of the Taiwan isolate O3D. The 5′-UTR (626 nt) and 3′-UTR (315 nt) showed 2.8% to 5% and 0.3% to 4.3% divergence respectively from those of other reported isolates (Table 2). Phylogenetic analysis showed strong homology (92.9% to 99.2%) between the VP1 gene from ZJ-V and that of 20 reference isolates (Table 2). The 21 DHV-1 sequences were clustered into 2 major genogroups (GⅠ and GⅡ, Fig. 2). GⅠ contained 11 tissue-adapted DHV-1 isolates, and can be further divided into 3 subgroups (S1 to S3). S1 includes RDL-62, R85952, ZJ-V, SH and H, while S2 contains 4 Chinese attenuated isolates (JX, C80, ZJ-A and ZJ-A2) which are very similar to one another. S3 contained a single isolate from Taiwan, O3D that was more distantly related, perhaps because of its geogra-phical location. Although ZJ-V was isolated in 2006 from a 1-week-old duckling, it had been passed 20 times in chicken embryos and grouped with the attenuated isolates. All field isolates that had not been multiplied in chicken embryo or cell lines were in GⅡ.

Table 2. Pair-wise comparison of the nucleotide and amino acid sequences of the VP1 genes from ZJ06 and selected DHV-1 isolates
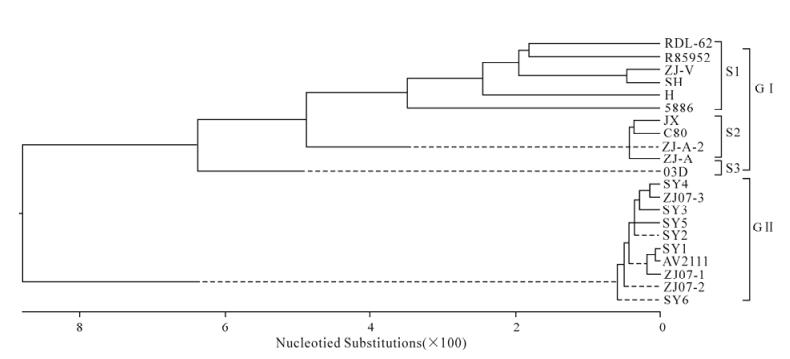
Figure 2. Phylogenetic tree of DHV-1 based on the sequences of VP1 gene. The phylogenetic relationships were estimated by using the Clustal V method. Reliability of the tree was assessed by bootstrap analysis with 1000 replicates.
Genetic organization of ZJ-V DHV-1
Nucleotide sequence similarities and divergence
-
Eleven oligonucleotide primers were used for amplification of the ZJ-V genome and 5 overlapping RT-PCR products were synthesized. Because these primers were designed according to the sequence of DHV-1 O3D, they were all located in the conserved regions of the DHV-1 genome. The 3′-terminal sequence of DHV-1 was amplified by the 3′-RACE method to get the true sequence of the ZJ-V isolate.
The 5′-UTR sequence has been determined in several isolates. The ZJ-V 5′-UTR consists of 626 nt and precedes the putative codon required for translation initiation. As expected, the 5′-and 3′-end sequences of the 5′-UTR are highly conserved between different isolates (94.2% to 99.5% homology). Computer-assisted prediction of the secondary structure of the ZJ-V 5′-UTR shows 4 separate functional domains in the structure (data not shown), supporting findings by Kim et al. that the ZJ-V structure correlates with the type Ⅱ IRES of picornaviruses and suggesting that the 5′-UTR of DHV-1 contains a type Ⅱ IRES.
The ZJ-V polyproteins contain 2 249 amino acids and share homology with other DHV-1 isolates. Sequence alignment revealed that the DHV-1 polyprotein contained a 3Cpro protease and potential proteolytic cleavage sites. The predicted S/H cleavage site between 2A2 and 2A3, in contrast, is not present in picornaviruses. In addition, there is only one copy of the 3B gene (the Vpg encoding region) in the DHV-1 genome, in contrast to other picornaviruses, such as FMDV, which have 3 copies (5). Although the 3B gene from ZJ-V shares low sequence identity with the FMDV 3B gene, the conserved tyrosine residue located at position 3 is still present. This residue is required for Vpg attachment to the 5′ uracil of the picornavirus genome (2, 15). An RGD motif is conserved among picornaviruses, which is important for virus-cell interactions (9). However the motif is not present in the ZJ-V VP1 gene, suggesting that DHV-1 attaches to the BHK-21 surface using one primary receptor.
Comparison between the complete nucleotide sequences revealed no striking differences between the ZJ-V, DRL-62, and 03D isolates. It demonstrates that VP1 is the most variable gene in the picornavirus genome (2), and phylogenetic analysis is usually undertaken based on alignment of the VP1 gene. To assess the genetic relationship between different DHV-1 isolates, the VP1 gene sequences were aligned using DNAstar, and a phylogenetic tree was construc-ted. The data confirmed strong homology between the VP1 gene from ZJ-V and the twenty reference isolates (Table 2). The maximum nucleotide difference was 8.1%, suggesting that there is limited variation among DHV-1 isolates.
The 3′-UTR of isolate ZJ-V is 315 nt, the largest in the family Picornaviridae, and shares high sequence homology (94.0 to 100%) with the reference isolates. The predicated secondary structure of ZJ-V suggests that it can form 4 putative stem-loop (SL) structures, SL1, SL2, SL3 and SL4 (data not shown). SL2, SL3, and SL4 are very stable. It is reported that the 3′-UTR is important for picornavirus replication (10, 3), but whether conservation of the 3′-UTR is related to its function requires further study.
This paper reported the complete genomic sequence of a DHV-1 isolate, ZJ-V, from Mainland China, which will be helpful for the taxonomy and molecular epidemiology study of DHV-1.

 DownLoad:
DownLoad: