











-
The detection of viral pathogens is an essential process in many areas. DNA microarray offers a highthroughput approach that has been proved to be a tool capable of detecting viral pathogens in a precise and sensitive way (17). However, some bottlenecks in the technique have been recognized, including nucleotide probe spotting/synthesis and probe design. In the production of microarrays, the density-versus-cost issue has been a major problem. Thus, for the highthroughput viral detection, new approaches are needed to reduce the oligo numbers without compromising the detection capacity and efficiency. The design of suitable sets of oligonucleotide probes is another important step in DNA microarray experiments. For viral detection, a number of groups have designed oligos for representative strains, but the remaining variants are beyond the capacity of the probes (2, 3, 11, 16). An alternative method involves the design of oligos within conserved regions to cover a wider range of variants (4, 8, 15). However, a significant amount of variants are still overlooked by this method, such as those with highly mutated sequences with insertions or deletions at the sites of probes (6). To avoid false-negative or ambiguous results, one can increase the number of oligos designed based on sequences in available databases to cover as many known variants as possible. However, this will lead to the higher probe numbers.
An effective oligo-probe set for viral detection should satisfy several criteria. The specificity is of the highest priority, and has been studied intensively. Using current computer programs (such as OligoArray 2.1) to design 50-mer probes for the type I human immunodeficiency virus (HIV-1) tat genes, we found that over 50% of the probes have the risk of cross-hybridization according to Kane's specificity criteria (7). The common problem using oligos longer than 50-mer is that the specificity is reduced with the increase in oligo length (12). Thus, a proportion of viral variants would not have specific probes if 50-mer or longer oligo designing methods were used. An alternative strategy is to design probes using the GoArrays approach. In this strategy, the oligonucleotide probe consists of the concatenation of two subsequences that are complementary to their target cDNA with an insertion of a short random linker, e.g. 6 bases, to facilitate the formation of the transcript loop (Fig. 1A). This strategy has been shown to broaden the range of oligo selection and increase the specificity and sensitivity (12). In addition to the specificity criteria, the oligo numbers should also be considered. When we tested the existing methods, such as to design oligos within the conserved regions or by the GoArrays approach, we found that all methods would require at least 1500 probes to cover the 1881 tat variants. Therefore, a strategy that circumvents the specificity and oligo number limitations is needed for the microarray-based viral detections.
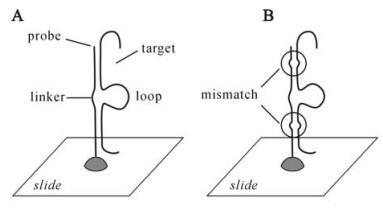
Figure 1. Schemes of existing and newly developed strategies for oligo design. A: The GoArrays strategy (12), in which the oligo-probe is composed of two disjointed subsequences from one coding segnence (CDS). Subsequences are concatenated via a short linker, e.g., 6 bases. The hybridization between the composite probe and the target induces the formation of a loop, as depicted. B: The modified strategy, in which the mismatch theory is adapted into the GoArrays method, i.e., non-perfect match (es) is incorporated in the concatenated subsequences that were selected from conserved regions.
In this paper, we describe a new strategy for oligonucleotide design aiming to cover all known variants for the viral species of interest with minimal oligo numbers. This approach was developed on the basis of the GoArrays approach (12) and the nonperfect match theory (9). Our results show that the oligo number is significantly reduced in comparison with using other published methods, while the specificity and hybridization efficiency remain intact. This approach could be generally applied to oligodesign or the development of low cost and high throughout viral detection arrays for clinical applications.
HTML
-
HIV-1 sequences corresponding to exon 1 of the tat gene for 2605 strains (by August, 2005) were downloaded from the LANL HIV Sequence Database (http://hiv-web.lanl.gov/content/hiv-db/mainpage.html). The aligned tat database (in FASTA format) was obtained by searching for all tat exon 1 at http://hiv-web.lanl.gov/components/hiv-db/combined_search_s_tree/search.html. Redundant sequences and sequences with bases other than A, T, G, C were removed. Specially, strain AF443106 was removed because it lacks a large portion of nucleotides. After the filtering steps, tat exon 1 sequences of 1881 strains were collected.
The BLAST program version 2.2 was downloaded from http://www.ncbi.nlm.nih.gov/BLAST/ download. shtml (1). The BioPerl (http://bio.perl.org/) Stand AloneBlast module was used to invoke, parse the result of the blastall program, and to check the specificity of probes designed. All BLAST programs were run using the parameter '-p n' to specify blast on nucleotides, and with default settings for other parameters. Unless indicated, the BLAST score of 32.2 for a 50-mer probe was used as specificity threshold. The human mRNA database used for BLAST to ensure specificity was downloaded from ftp://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/mRNA_Prot/.
OligoArray 2.1 (13) was downloaded from http://berry.engin.umich.edu/oligoarray2_1/; the required OligoArrayAux was downloaded from http://www.bioinfo.rpi.edu/applications/hybrid/OligoArrayAux.php. We set the oligo length 50-mer; maximum oligo number 1; other parameters were set as default. GoArrays (12) was downloaded from http://www.isima.fr/bioinfo/goarrays/, and run following the authors instructions. Maximum length for identity was set as 16; subsequence length was set to 22; other parameters were as default.
-
The Tm value was calculated by the program downloaded from DINAMelt web server (10) (http://www.bioinfo.rpi.edu/applications/hybrid/hybrid2.php) and run locally as instructed. The initial concentrations of two strands were set as 10-7mol/L; [Na+] was set as 0.5mol/L; other parameters were set as default (see'specificity evaluation' for explanation of these choices).
-
The free energy (dG) of hybridization was calculated by Mfold web server (18) (http://www.bioinfo.rpi.edu/applications/mfold/). To predict the dG of structure stability, the probe sequence was fixed whereas the target sequences varied. Point mutation(s) in the matched regions was generated by replacing perfectly matched pair(s) with one of the other three non-matching nucleotides.
-
A conserved region of certain length of nucleotides is determined by the variety within each window, which shifts from the beginning to the end of the exon by a fixed offset of 5 nt (slide the window along the sequence in 5 nt steps) for each strain in the database. A window with a higher number of unrepeated subsequences is considered to be of higher variety and less conserved. Each sequence selected from a conserved region was subjected to the BLAST program to confirm its specificity as to be used as a probe.
Database and programs
Melting temperature (Tm) calculation
Free energy and hybridization structure prediction
Determination of conserved regions
-
We first tested whether concatenating two shorter conserved sequences by the GoArrays strategy could be a practicable way to reduce oligo numbers. We obtained several conserved regions (22-bases each) of the HIV-1 tat gene as targets for design, and developed a corresponding program (see 'dynamic programming' below) to link two subsequences selected from the conserved region pools. 660 specific probes were deduced to cover all 1881 variants, suggesting that selecting shorter conserved oligos can help to reduce oligo numbers.
Incorporating appropriate mismatches into oligoprobes could be another way to reduce probe numbers, while the feasibility of non-perfectly matched probe design has been highlighted by other studies (9). To study the capability of mismatches in reducing oligo numbers, we first chose a short conserved region (e.g., the 22-base sequences beginning at position 106 in the tat exon 1 alignment), then calculated the amount of oligos and their inclusive strains at given number of mismatches. As shown in Fig. 2, the more mismatches that were incorporated, the fewer the number of oligos that were required while maintaining the same coverage. Choosing other regions and/or other oligo length (>20-mer) showed the same trend (data not shown). The results demonstrate that introducing non-perfectly matched probes can help to reduce the number of oligos.
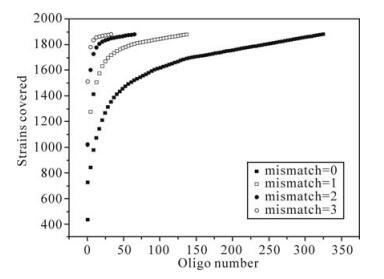
Figure 2. The relationship between calculated strain numbers and oligo numbers with different mismatches. The region used for calculation is 22-bases, beginning at the position 106 in the tat exon 1 alignment. Curves representing the number of mismatches are indicated in the inset. To cover all 1881 strains, there are 326, 138, 65 and 34 oligos corresponding to 0, 1, 2, and 3 mismatches, respectively.
The above approaches to reduce oligo numbers prompted us to develop a new strategy based on the GoArrays method and the mismatch theory in order to get the minimal number of probes. The scheme of our newly developed strategy is shown in Fig. 1B. Here, designed mismatches are incorporated in the matched subsequences selected from the conserved regions. In the following results, we describe the program to optimize oligo number and specificity, and then validate the related parameters to demonstrate the feasibility of the strategy.
-
Prior to optimizing the oligo numbers, we determined the conserved regions for subsequence pools, and then optimized the combination of linkages between every two pools using a dynamic programming algorithm in order to obtain the maximal coverage while using the minimal number of concatenated probes. Here we define a 'linkage' as a concatenation of two subsequences from two separate pools, whereas 'combination' as a set containing one or more linkages selected from the set of all possible linkages (scheme seen in Fig. 3).
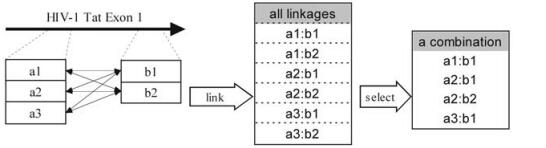
Figure 3. Scheme of linkage and combination. (Left) The axis indicates the HIV-1 tat 1st exon used for design. Open boxes represent different oligos within two regions. It is important to note that there is more than one way to link the subsequences. (Middle) A list shows all ways of linkages in the given situation. (Right) An instance of a combination, which is a subset of all linkages.
The first step was selection of two regions for linking. By determination of conserved regions (described above in Methods), we were able to obtain several candidates (the length of each subsequence is 22 bases, see 'specificity evaluation' below). We then selected these regions from aligned HIV-1 tat database to get the subsequence pools. Next, redundant sequences in each pool were removed, and the rest were subsequently filtered to ensure specificity by the BLAST program. Thus, all eligible sequences in each pool were identified and saved.
The next step was to optimize the combination of linkages using dynamic programming in order to obtain the maximal coverage while using the minimal number of probes. If the oligos in two subsequence pools are to be linked, the length of the linkers should fall between 3 and 100 bases, according to the restrictions by GoArrays strategy (12). Meanwhile, a probe must satisfy the condition that no more than 1 mismatch in each subsequence (see 'specificity evaluation' below). To get an optimized (n+1)-linkage combination, we need an optimized n-linkage combination and a selected linkage, which, after joining in the n-linkage combination, makes the new (n+1)-linkage combination cover the maximal number of targets (Fig. 4). This dynamic algorithm ensures that the coverage at each step is maximized, and that the number of probes to cover the corresponding variants is minimized. The best resulting combination can cover 1875 out of 1881 strains with only 272 probes. The remaining 6 undetectable strains resulted from the preparation step were saved in a file for future use. Therefore, our strategy greatly reduces the number of oligos to 272, from over 1800 by GoArrays approach (12) or over 1500 by 50-mer or longer oligos with conserved region method (15).
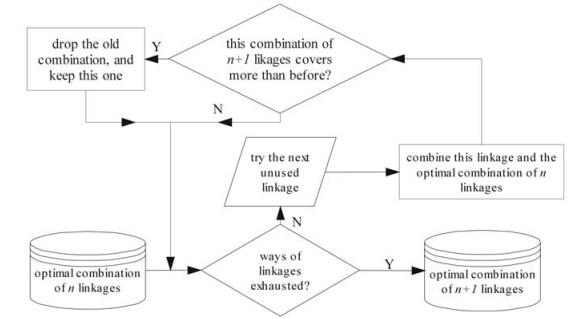
Figure 4. Flowchart of optimizing a combination by dynamic programming (a single step of recurrence). A set of optimal n-linkage combination is input in the recurrence, and all possible ways of linkages are exhausted to find a best linkage in order to get the optimal (n+1)-linkage combination. Such steps are repeated to complete the whole step. See text for more details.
A random linker of 6 bases was then concatenated between each subsequence. The specificity of every linked probe was again confirmed by the BLAST program.
-
The melting temperature (Tm) has been widely used in simulating hybridization conditions as well as cross-hybridization (9). In this study, we used the DINAMelt web server to calculate the Tm and the melting curves (presented by hybridization rate of targets/non-targets and their probes as a function of temperature). The deviations of the model were evaluated first. We compared the calculated Tm with the experimental dissociation temperature (Td) from two groups of sequences (55 ones in total) that contain single point mutation in different site, by Urakawa and colleagues (14). The linear correlation coefficients R of the two groups were 0.79 and 0.88. Using conditions described in 'Materials and Methods', as commonly used in software and experiments, the correlation coefficients were found to be better than those obtained with default parameters (R equals 0.78 and 0.84 respectively). Although the calculated Tm are about 20℃ higher than the Td data, the absolute value is less essential than the overall correlation, since we can simply reduce the 20℃ for each datum. All Tm were therefore calculated by the DINAMelt with the modified parameters.
The length of the subsequence and the amount of mismatch incorporated in the oligo are directly related to the specificity of the probe. Longer oligos and fewer mismatches contribute to the specificity, whereas shorter oligos and more mismatches have the advantage in reducing oligo numbers. To balance the relative merits of these two extremes, we sought a combination of these two requirements that maintained specificity with a manageable number of oligos. After a series of calculation and comparison (data not shown), we found an optimized parameter: 22-bases for subsequence length and no more than one mismatch for each subsequence (scheme shown in Fig. 1B). This parameter ensures specificity (as measured by the hybridization rate of target-hybridization and cross-hybridizations described below) and minimized oligo numbers (as compared and calculated by dynamic programming described above).
The specificity evaluation was here presented. Among the designed probes, the probe with the lowest specificity was identified from the BLAST results. This probe hits many unrelated sequences in the specificity database. Based on the calculated melting curves of this probe with the various sequences (targets and non-targets), we then generated the melting profiles for overall hybridization (data not shown). Using these profiles, we were able to predict the hybridization rate of this probe with its targets or non-targets at given temperature. The criteria for a specific hybridization, as commonly used by previous studies (9), are that hybridization rate for target sequences is over 95%, and for non-target sequences, less than 15%. To simplify the scenario, we picked 64 to distinguish the specific an ℃ d non-specific hybridization. The calculated Tm and hybridization rate of selected sequences are shown in Table 1. The extremity of undesirable condition is represented by the target AF325758 and the non-target Cross 5. AF325758 has the lowest target-hybridization rate among all the targets, while Cross 5 has the highest cross-hybridization rate among all the non-targets. As seen in Table 1, the hybridization rate of AF325758 and Cross 5 still satisfies the specificity criteria mentioned, indicating the specificity of the tested probes. Since the probe used in above process was of the lowest specificity, it is evident that all the other designed probes would meet the criteria as defined above.

Table 1. Calculated hybridization Tm and corresponding hybridization rate of selected sequences at 64℃.
-
The stability of the hybridization structure is of the most important aspect in hybridization efficiency and sensitivity for DNA microarrays. In our design, the influential factors affecting stability are the mis-matches and the internal loop. We therefore investigated these two quantities.
To examine the influence of mismatch on the stability of hybridization, the free energy (dG) was calculated using the same probe and mismatch parameters determined above. To test a more general situation, we deliberately introduced series of nontargeting sequences with satisfied mismatch threshold (referred to below as artificial sequences).The change in dG was measured by ΔdG (ΔdG = dG mismatched -dG perfect matched). For the actual targets, a single point mutation yields ΔdG 1.5~4.5kcal/mol, whereas the maximum ΔdG can reach to 5.5 kcal/mol for artificial sequences, as shown in Fig. 5. For double point mutations in the investigated probe (one point mutation in each subsequence), the ΔdG is 3~9 kcal/ mol for actual targets, while it can be as high as 11kcal/mol for artificial sequences.
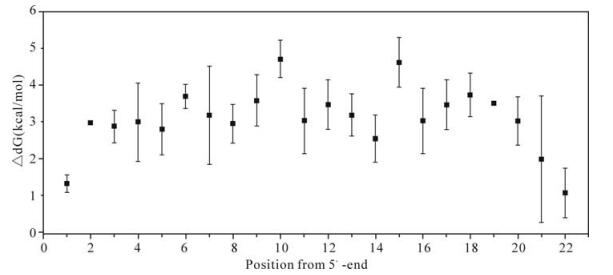
Figure 5. Calculated ΔdG for a 22-mer oligo with nucleotide mutation. Average ΔdG values of each mutation at different positions are shown with standard deviations. Probe sequence: GCTTCGTCGCTGTCTCCGCTTCgacactAAGCAAACTAAACAGTGGTA GC; lower case letters represent linker sequence; bold letters represent the subsequence investigated in the graph. Results in another subsequence are similar to this graph (data not shown).
To examine the influence of internal loop on the stability, we generated one artificial 'open loop', wherein none of the nucleic acids could match. This represents the extreme scenario in which the hybridization stability is ruined. The change in dG is still measured by ΔdG (ΔdG = dG modified -dG perfect matched). We found that the greatest ΔdG could reach as high as 6.5kcal/mol for artificial sequences. In contrast, the ΔdG was 0~4.1kcal/mol for actual targets. The influence of loop on the stability is therefore a less important factor compared with that of mismatches in subsequences.
As far as both mismatches and loop are concerned, the dG for actual targets is -48.5 to -54.2 kcal/mol, whereas the greatest dG for artificial sequences is –40.1 kcal/mol. In the GoArrays strategy, a 20-basesubsequence probe with the dG around -40 kcal/mol can hybridize even more efficiently than the classical 50-mer probes (12). The thermodynamic data show that the dG of our probes is much lower than, or comparable with -40kcal/mol, indicating the intact hybridization stability of those probes designed by our strategy.
-
Other parameters affecting hybridization, such as Tm range, secondary structure and prohibited sequences, were considered (data not shown). Ineligible probes were excluded and re-designed, and 9 additional probes were added. Thus, the total probe number to sufficiently detect all 1881 HIV-1 tat variants should be 281 (272+9). In conclusion, the oligo number in our design strategy is significantly reduced while the specificity and sensitivity of the probes are better than, or comparable with other reported studies mentioned.
In summary, our strategy for designing viral detection probes can be summarized by the following four steps. (1) Determination of suitable 22-mer regions which are conserved and specific. Sequences in two of the selected regions will be used as subsequence candidates. (2) Pre-filtering of subsequence candidates for each of the selected regions. Identical sequence from different strains should appear only once; prohibited sequences and sequences with BLAST scores higher than 32.2 (for 50-mer probes) should be eliminated. (3) Linkage of subsequences into probes, using the 'dynamic programming' described above (the computer program implementing this algorithm step is freely available on request: zhangchn2004@gmail.com). (4) Examination of the designed probes for Tm consistency, secondary structures, and other parameters.
Strategies to reduce the number of oligos
Optimizing oligo numbers by dynamic programming
In silico evaluation of specificity
Thermodynamic validation of hybridization stability
Further verification of the probe set and the guidelines for viral probe design
-
During our in silico study, we found that existing specificity criteria for oligonucleotides (7, 13) can be further improved. More recently, He Z. and colleagues proposed an improved criteria which combines identity, stretch and free energy (5). However, we noticed that combining even these three parameters is still not sufficient to ensure specificity, because we were able to create sequences that satisfy all three conditions but still produce non-specific cross-hybridization regardless of hybridization temperature (our unpublished data). Therefore, other thermodynamic parameters, such as Tm and the hybridization rate, should be investigated.
An requirement for DNA microarrays in virological applications is that the methodology could detect unknown variants in the given viral species as well as previously identified variants (15). However the rapid point mutations and segment reassortment of viruses have been a challenge for probe design. When we updated the database in October 2005, we found that there were another 58 new variants posted for HIV-1 tat in addition to the database in August that year. Promisingly, 46 of the 58 new variants can be covered in the 281 probe set as we have deduced, indicating the powerful detection capacity of the probe design.
Further considerations should also be noted. First, as to the probe number, there is still room for improvement. We found that a number of probes contribute only one target in later steps of recurrence during the dynamic programming. To synchronously perform linking combination at more than one pair of regions could be one solution. Another plausible way is to design artificial sequences that do not exist in the subsequence pool but satisfy the mismatch threshold for the targets in later steps. Second, although our calculation tools are currently most updated and the in silico data are statistically reliable, there are still certain deviations compared with experimental results. Finally, our strategy and the validations are based on theoretical model, therefore further experimental verifications are imperative.

 DownLoad:
DownLoad: