











-
Tracing influenza viruses' evolution has been the subject of much research relevant to influenza viruses and frequent reassor-tment events for both avian and human influenza viruses have been detected using phylogenetic methods (6, 13). For instance, several genotypes of H5N1 avian influenza viruses have been detected in the past five years, and these have been designated A, B, C, D, E, G, V, W, X, Y, Z, Z+ and so on (4, 5, 11, 13). Viruses of genotype A, B, C, D, E and F were also identified for the H9N2 subtype (1). There is no denying that these studies revealed differences among viruses. However, all of the differences were principally at the DNA level and genetic information at the protein level was not fully utilized. Therefore, phylogenetic trees sometimes could not provide subtle differences among viral genes.
To better make use of the information at the protein level, molecular characterization analyses and reverse genetics techniques have been performed to help find the key sites relevent to pathogenicity, virulence and even host selection of influenza viruses (15, 16) etc. Up to date, some positions playing important roles in viral genomes have been found, such as the con-necting peptide sites in HA, Lys-627 in the PB2 fragment (7, 10) and so on. Thus, molecular characteri-zation analysis does have advantages in seeking single amino acid and short peptide mutations. However, it is difficult for it to integrate all these genetic information as a whole to find genes that co-reassort or proteins displaying compensatory mutations. To this end, Obenauer et al. introduced a proteotyping method to visualize unique amino acid signatures (proteotypes) (17). This method was able to identify co-reassorting genes, 50+ protein-protein pairs, virus "families" that share specific combination of genes and proteins exhibiting compensatory mutations (8).
Neuraminidase (NA) is a surface protein that cleaves sialic acid from virus and host cell glycocon-jugates at the end of the virus life cycle to allow mature virions to be released (25). Phylogenetic studies have revealed that the H5N1 avian influenza viruses of China were divided into three lineages according to the NA gene tree, with one lineage (Ⅰ) possessing a 19-aa deletion in the stalk of NA, one lineage (Ⅱ) without deletion, and one lineage (Ⅲ) with a 20-aa deletion (9, 24). Viruses of genotypes A, G, X, Y, Z and ShanTou3-like (ST3-like) belonged to group Ⅲ, while B, C, D, E, W, Z+, ST1-like and ST2-like isolates belonged to group Ⅱ and HK/156/97 was placed into group Ⅰ.
In this paper, we took NA gene fragments of some H5N1 influenza viruses isolated from mainland China, Hong Kong Special Administration Region (SAR) and Southern Asia as an example to illustrate how the proteotyping method worked.
HTML
-
Our dataset included the NA gene segments of typical H5N1 avian influenza viruses of the known genotypes isolated from mainland China, Hong Kong SAR and Southeast Asia. In addition, some isolates from human were also included to assist our analysis. Parrot/Ulster/73 was designated as an outgroup to root the tree. All nucleotide sequences were obtained directly from GenBank.
The first step in proteotyping was similar to that of normal phylogenetic analysis. Multiple sequence alignment was performed with ClustalX 1.81 (23) and the alignment parameters were set to default. To estimate the trees accurately, MrBayes, version 3.0b4 was used to construct the NA gene tree (19). Four Markov chains were run for two million generations and sampled every 100 generations to yield a posterior probability distribution of 20 000 trees. After elimi-nating the first 5 000 trees as burn-in, a 50% majority-rule consensus tree was constructed. Bayesian Posterior Probability (BPP) was used to assess the support for the recovered clades, given the aligned sequence data. A six parameter substitution model (General Time Reversible) was used with a gamma rate parameter allowing site variation. It should be noted that besides Bayesian, other trees search methods can also be used.
In the second step, DNA data were translated to their protein sequences accordingly by using Mega 3 (12). Alternatively, protein sequences could be downloaded directly from GenBank. After that, protein sequences were aligned using ClustalW included in Mega 3. The protein alignment was then re-sorted according to the sequence order displayed by the tree. Consequently, a so-called "clade-guided" sequence alignment was produced by assigning a unique color to each kind of amino acid. It also should be noted that leading and trailing gaps were generally artifacts of aligning sequences with different 5' and 3' termini (22) and were set to white. The remaining gaps were set to black in order to highlight the real amino acid deletions.
Thirdly, a consensus sequence was calculated for the alignment. All the consensus amino acids were set to white to match the background color so that only non-consensus sites were visible. Obenauer et al. proposed a residue occur more than any other residue to be the consensus (17). However, by our method, all the residues would be displayed if no residue occurred more than 50% in the column. The remaining residues were used to define the proteotype according to the numbers of variable amino acids among proteins.
Finally, the proteotypes of NA proteins of the representative H5N1 viruses were determined mainly based on the amino acid differences among protein sequences and position information of the sequences on the tree. After the proteotypes are determined, serial numbers will be assigned starting at the top downwards for each proteotype and these numbers would be summarized into a table 1. At the same time, unique amino acids were sought from the NA proteotypes.
-
The NA gene tree was mostly divided into two major lineages with a small branch out of them (Fig. 1). One major lineage involved viruses of genotype A, G, X, Y, Z, while the other involved genotype B, C, D, E, W, Z+.
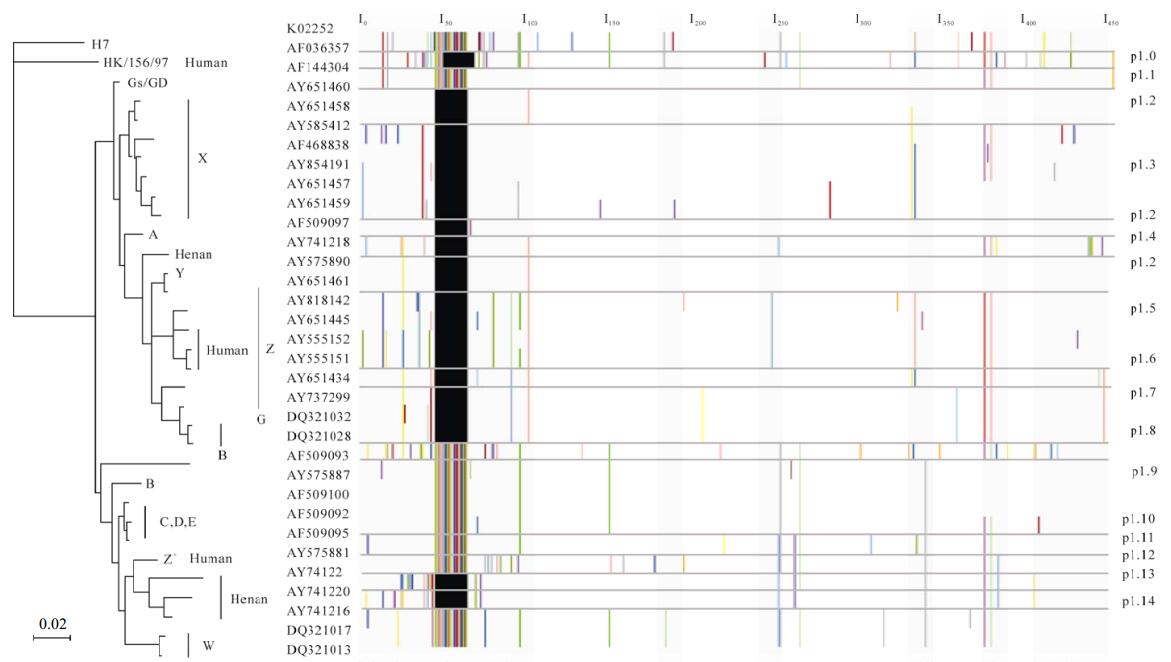
Figure 1. Proteotypes for NA genes/proteins of some H5N1 avian and human influenza viruses. Phylogenetic analysis was based on nucleotides 20-1426 ( 1,407 bp) of the NA gene and the tree was rooted to K02252 (A/Parrot/Ulstcer/73. H7N1). Following the GenBank accession numbers there was the corresponding genotype or host information of these viruses. Scale bar. 0.02 nucleotide change per site. The left column was the GenBank accession numbers of the representative H5N1 avian and human influenza viruses. The protein alignment was adjusted according to the sequence orders of the viruses in the NA gene tree. The right column was the serial numbers designated to the NA proteotypes respectively.
-
Proteotypes of NA proteins supported the phy-logeny revealed by the NA gene tree (Fig. 1). However, there were some differences between the results of the phylogenetic and proteotyping analyses. First of all, the proteotyping analysis displayed protein differences within the lineage and even within the genotype. For example, the differences of NA proteins among the viruses of the X genotype were observed. Likewise, our results indicated that the Z genotype viruses might be further divided into more proteotypes (Fig. 1). Secondly, some proteotypes might involve more than one genotype. For instance, some viruses of genotype X, A, Y and Z were defined as the same proteotype -p1.2 (Fig. 1, Table 1). Finally, some co-variable amino acids that might be potentially important to maintain the advanced structures and functions of the proteins were found. Particularly, it was possible that Thr17, Lys64, Asn75, His233 and Ser320 co-evolved in the NA proteins of some Southeast Asia isolates of Genotype Z (Fig.1). It also suggested some sites of viruses of genotype W might evolve with each other (Fig. 1).

Table 1. Some representative H5N1 avian and human influenza viruses and their corresponding NA proteotypes
Bayesian Analysis
Proteotyping Analysis
-
Proteotyping is a recently introduced method akin to genotyping at the DNA level, but wichit additionally captures the variability of proteins as they occur in populations and change over time (20). Using this method to help find the proteins related to diseases and study the changes of these proteins both in healthy and morbid situations has been reported (2, 3, 26). It has also been used as a tool to study developmental lesions (21). Some researchers even used it to link genotype and phenotype of some diseases (14). In these studies, the proteotyping processes were often fulfilled by the assistance of mass spectrum (MS) (18, 20).
Method has also been reported to be used to study influenza virus evolution at the protein level (17). In this study, it is have modified and has some particular characteristics. First of all, Proteotyping analysis is principally sequence-based, and therefore it can be completed without MS data. Secondly, as mentioned in the method section, the protein sequence alignment has been changed to clade-guided rather than normal multiple sequence alignment. Thirdly, for influenza viruses, genotype is only determined by the whole genome rather than single or few genes of it. In contrast, the proteotypes of the viruses can be determined for both each gene of the virus and the whole genome. In fact, integrating all the eight proteotypes determined for each gene segment, one can ascertain the proteotype of the whole genome like what have done to define a genotype of an influenza virus. Fourthly, the serial numbers designated to the proteotypes of the same viruses may be different because the serial numbers are decided both by the sample size into analysis and by the positions of the viruses in the gene tree. At last, the proteotype can also be linked to genotype. In fact, information at the genotype level is helpful to define the proteotype.
Bayesian analysis in this paper confirmed the previously constructed topology (9, 24). However, differences between the results from phylogenetic and proteotyping analyses proved that the proteotyping method had a higher resolution and was able to mine more subtle differences among viruses. The specific amino acids found by the proteotyping method could be further analyzed by other bioinformatics techniques or reverse genetic techniques to study their potential biological functions. However, only the proteotypes of NA proteins were determined here (Table 1). If the proteotypes of all the eight proteins of the influenza viruses were identified, proteins co-reassorting or showing compensatory mutations could be detected (17).
Unlike the consensus definition proposed by Obe-nauer et al.(17), we suggest all the residues should be displayed if no residue occurs more than 50% in the column. If none of them are occurring more than 50%, this may be an indication that this site is super variable. Although a super variable site suggests weak selective pressure and absence of biological function, if these variable sites were neglected, it is difficult to find sites that might co-reassort and they would be hidden subjectively. It is likely that these coreassorting sites might be related to the function of the protein. Therefore, hiding the residue taking up less than 50% in the column might lose potentially important information.
It should be also mentioned that there is no general criterion availabel to guide the definition of a pro-teotype. If it is defined arbitrarily, potentially, useful information might be hidden by the noise. However, the number of different amino acids among proteins and the positions of the viruses in the gene tree may be helpful to distinguish different proteotypes, but in some cases this may not be sufficient. Additional factors should be also taken into account such as serotype, subtype, genotype, host, collection time and natural selection pressure. In particular, sample size is also an important factor that should not be ignored. However, in this paper, we mainly introduced the proteotyping method and therefore only a sample of small size was used and the proteotypes were designated mostly by the numbers of different amino acids of NA proteins. Consequently, the proteotypes designated here were not strict.
To sum up, proteotyping method is a useful tool for studying virus evolution at the protein level. It also can be applied to other viruses, especially to viruses with segmented genomes.

 DownLoad:
DownLoad: