











-
An epitope, or antigenic determinant, is the part of a macromolecular complex which is recognized by the immune system and is classified according to its targeting receptor. T cell epitopes, as presented in the major histocompatibility complex (MHC) molecule, are antigenic peptide strings recognized by T cells receptors. MHCⅠ molecules present endogenous antigens while MHCⅡ molecules present exogenous antigens. The MHCⅠ molecule binds to a peptide of approximate 9 amino acids in length within a closed groove. In contrast, because the antigen-binding groove is open at both ends, the MHCⅡ molecules can present much longer peptides, generally varying from 12 to 25 amino acids, nine of which occupy the binding groove. This difference between MHCⅠ and MHCⅡ is very important for the development of distinct prediction algorithms. B cell epitopes, which are recognized by antibodies or B cells, are divided into a series of continuous linear epitopes and discon-tinuous conformational epitopes. The conformational epitopes that comprise the major B cell epitopes can be considered in terms of the three-dimensional (3D) surface features they contribute to the antigenic molecules, whereas the linear epitopes are defined by the amino acid sequence rather than by their 3D shape.
Given their impact on global health, much effort has been expended into elucidating the mechanisms utilized by viruses in order to elude the host immune system and development of prophylatic and ther-apeutic vaccines. Traditional vaccines are based on inactivated or attenuated pathogens. While they play an important role in the protection of infectious diseases, these vaccines have biohazard risk since they may infect the recipients. Compared to traditional vaccines design, epitope-based vaccines provide a safer approach as they consist of rationally designed protective epitopes, which are able to stimulate effective immune responses whilst avoiding potentially hazardous and undesirable side effects. Nevertheless, the identification and selection of suitable epitopes is a time-consuming and expensive work requiring careful experimental screening. Hence, there is a need for developing a quicker and cheaper strategy to address this bottleneck. One option is to use computational predictions and there are many bioinformatics tools available. Also, there are several public accessible epitope-associated databases, such as IEDB (The Immune Epitope Database and Analysis Resouces; http://www.immuneepitope.org) which are further sources of information. The IEDB database is a repository of curated empirical epitopes data, comprising both positive and negative subset, which can be used as a benchmark. Since these types of computational tools have been incorporated into the vaccine screening pipeline, epitope based vaccine design has become significantly more efficient. Even though the effectiveness of these prediction tools are limited by their accuracy, it is likely that, with the increasing amounts of experimental data such as genome sequences and protein structures, and the development of new and more advanced algorithms, more powerful and efficient virus epitope prediction tools will be available in the near future.
HTML
-
The underlying assumption in these methods is that some evolutionary relationship exists amongst groups of viruses and sequence analysis can discover conserved patterns within these groups. The manner in which these patterns are identified depends on the class of epitope and the particular software. Both T cells and B cells epitopes prediction are essential for epitope-based or epitope-driven vaccine design and a variety of prediction algorithms have been developed based on the antigen's primary amino acid sequence, 3D structure or other protein characteristics such as hydrophilicity, accessibility and flexibility. Several epitopes prediction softwares are currently available (Table 1).

Table 1. The list of currently available epitope prediction softwares
The tools listed above performed differently in terms of accuracy, specificity and sensitivity. The predictive results comprise true positive (TP), false positive (FP), true negative (TN) and false negative (FN). Generally, the accuracy which measures the ratio of correct predictions comprising true positive and true negative by (TP+TN)/(TP+FP+TN+FN), is the major criterion for assessing the quality of a predictive model. A predictive model with higher accuracy is often better than the lower one. Also, the sensitivity which measures the ratio of true positive by TP/(TP+FN), the specificity which measures the ratio of true negative by TN/(TN+FP) are important criterions. The accuracy is increased by increasing simultaneously both sensitivity and specificity, or by increasing either. Theoretically, an optimal prediction can reach 100% sensitivity and 100% specificity, which means the 100% accuracy; while in practice, it is impossible to achieve such a perfect prediction, there is usually a trade-off between the measures. Usually, the machine-learning based algorithms perform better than the motif-based algorithms in T cell epitopes prediction and linear B cell epitopes prediction with a higher accuracy. For example, SVMHC is more accurate than SYFPEITH (T cell epitopes prediction); while ABCpred has a significant increase in accuracy compared to Bcepred (linear B cell epitopes prediction). The conformational B cell epitopes prediction algorithms are based on 3D structure, and they have different performance. For instance, CEP predict more accurate than Discotope. However, there are no perfect algorithms, every tools has its strengths and weaknesses. In general, in practice several tools should be combined for epitopes prediction.
-
Given the different properties of the epitopes, prediction tools are generally MHCⅠ or MHCⅡ specific, although some software predicts both. The first prediction tools were motif-based algorithms, which predicted T cell epitopes by searching experimentally verified MHC binding motif sequences identified from affinity data. Several tools come under this classification, such as SYFPEITHI[17], ProPred[20]. Nevertheless, one of the drawbacks of these motif-based methods is that novel motifs are not recognized and so large numbers of false positive and false negatives can be generated. More recently, more sophisticated methods using, various machine learning based algorithms have been developed based on support vector machines (SVM)[6, 7], hidden Markov models (HMM)[16]and artificial neural networks (ANN)[3]Compared with motif-based algorithm, these machine learning algorithms are more accurate and efficient, especially when they were used in complicated pattern recognition.
SVM and ANN based algorithms work by using a positive set of experimentally verified epitope sequences and a second set of negative sequences to train the system to classify query sequences as belonging to one of these two classes. These is achieved by defining a set of N descriptive features for these sequences (such as nucleotide or dinucleotide sequence composition) then training the system against these positive and negative datasets. For a model defined by two features, this would involve trying to find a line that divides the two datasets in a two dimensional plot. More generally, this extends to attempting to find the N-1 dimensional hyperplane that distinguishes the two sets in the N dimensions of the feature space. The reliability of the trained model is investigated by cross validation, a process in which the training is subdivided into smaller sets and the system is retrained. Finally, the accuracy of the model is tested with the training dataset.
HMM based prediction methods work by representing the difference between a experimentally verified epitope and a query sequence as a statistical process. For example, a single base difference between the two sequences would require a single state change in the form of a single mutation. The probability of the state changed is estimated from likelihood of the change in the experimental sequences. In this way, query sequences which are more similar to known experimental sequences require few state changes and have a higher probability of classification as possible epitopes.
In brief, the steps of machine learning-based epitopes prediction algorithms are: 1) Data collection and processing; 2) Model building; 3) Parameter optimization; 4) Epitopes prediction. This is summarized in Fig. 1.
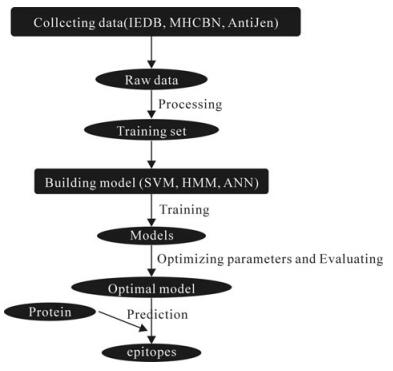
Figure 1. The flowchart of machine learning-based epitopes prediction algorithms
-
Compared to T cell epitopes prediction algorithms, the B cell epitope prediction is more complicated, especially for the conformational B cell epitopes because, in addition to the sequence composition, the 3D-structure of protein must also be considered.
The development of B cell epitopes prediction algorithms has been less successful compared to T cell epitope prediction, especially in accuracy. There are several reasons for this. For instance, the majority of B cell epitopes are discontinuous so that it is hard to determine the relevant amino acids and the dis-tribution of the antigen surface. Moreover, much of the experimental data which the prediction algorithms are based on are still controversial because of the poorly understood recognition properties of crossreactive antibodies [1, 4]. Nevertheless, in spite of these difficulties, there are several methods available for B cell epitope prediction for both linear and confor-mational epitopes. The prediction algorithms for linear B cell epitopes are similar to the T cell's. Similarly, the accuracy of primary sequence-based algorithms is low[11], and modified algorithms based on machine learning were subsequently developed, such as ABCpred[18] and BepiPred[14] with significant improvements in accuracy. Prediction algorithms for conformational B cell epitopes based on 3D structure are also available owing to the ever-increasing 3D structure of antigen-antibody complex data. Some online prediction servers based on this algorithm are accessible, for example DiscoTope[9] and CEP (http://bioinfo.ernet.in/cep.htm) [13]. These methods make use of information carried in the structure of antibodies against proteins of interest to reveal the 3D folding of target proteins.
T Cell Epitopes
B Cell Epitopes
-
The methods described above have been widely used in virus epitopes prediction, and aided the vaccine development process. The potential of these prediction tools is highlighted by summarizing some of the more significant results.
Human metapneumovirus (hMPV) cytotoxic T-lymphocyte (CTL) epitopes were predicted by combining SYFPEITHI (with PAProc) and ProPred1. When tested experimentally, some of these epitopes were able to stimulate a strong immune response, and vaccination with hMPV CTL epitopes could protect the hMPV-challenged mice. These results demon-strated the efficacy of an hMPV CTL epitope vaccine in the control of hMPV infection in mice for the first time[10]. The MHCⅠ T cell epitopes of porcine reproductive and respiratory syndrome virus (PRRSV) glycoproteins4 (GP4), 5 (GP5) and nucleocapsid were also predicted by SYFPEITHY and IEDB analysis while the MHCⅡepitopes were predicted by ProPred. These prediction were subsequently applied in in vitro and in vivo experiments and the results showed that some of these epitopes could provoke an immune response in pigs against PRRSV[5]. More recently, six HLA-A2 restricted CTL candidate epitopes of LMP2A (latent membrane protein 2A) of EBV (Epstein-Barr virus) were predicted by a combination of the SYFPEITHI, NetMHC and MHCPred[8] packages and three of six peptides were identified as LMP2A-specific CD8+ T-cell epitopes with functional experiments in vitro. It suggests that these three epitopes are good candidates for developing of a vaccine against EBV-correlative nasopharyngeal carcinoma[21]. Finally, eight candidate HLA-A*0201-restricted epitopes of the spike protein of SARS-CoV were predicted by SYFPEITHI and ProPred1 and four of the eight were tested by HLA-A*0201 binding assays. Among these, one peptide (Sp8) induced specific CTLs both in vitro (Peripheral blood lymphocytes of healthy HLA-A2+ donors) and in vivo (HLA-A2.1/Kb transgenic mice). Thus, the Sp8 epitope should help in improving the understanding of the mechanisms of virus control and immunopat-hology in SARS-CoV infection[15]. In addition to these examples, many other important virus epitopes have been predicted and verified, such as HIV[12, 19], Influenza A virus[2] and Foot-and-mouth disease virus[22].
-
Many in silico epitope prediction tools are available, which can complement experimental methods for epitope identification. Since these methods are performed in silico, they can be used as an initial screening step to identify targets of interest for more detailed experimental studies. Such an approach is more efficient in terms of time and cost. However, when using such an approach, it is important to recognize the limitations of current software tools; it is impossible to develop an exact algorithm, owing to the incomplete knowledge about the immune response and these methods are an approximation at best. Moreover, the prediction results produced by different tools may have distinct differences and multiple tools should be used to obtain a consensus result. Also the present tools need to be modified based on the increasing experimental data. Clearly, the challenge to develop novel, more systematic and accurate algorithms remains. However, the availability of ever-increasing amounts of virus genomic and proteomic information, coupled with advances in the development of new algorithms for sequence analysis will result in more effective and accurate tools for epitope prediction.

 DownLoad:
DownLoad: