











-
Acquired immunodeficiency syndrome (AIDS) was first reported in 1981. Today, an estimated > 33 million people globally are infected and living with the etiological agent of AIDS, Human immunodeficiency virus type 1 (HIV-1), a member of the Retroviridae family of viruses, Lentivirus genu; and approximately 25 million individuals have died from AIDS [22]. Although there are now more than 30 anti-HIV drugs developed, none can eliminate the virus completely and no prophylactic vaccines are currently available. Therefore, HIV continues to represent a significant public health problem that mandates intensified efforts and investment in both clinical management and research.
Mitogen-activated protein kinase (MAPK) pathways consist of a group of serine-threonine kinases and link extracellular signals to the machinery that controls fundamental cellular processes such as growth, pro-liferation, differentiation, migration, and apoptosis [10]. MAPK pathways contain a three-tier kinase module, in which a MAPK is activated upon phosphorylation by a mitogen-activated protein kinase kinase (MAPKK), which in turn is activated when phosphorylated by a mitogen-activated protein kinase kinase kinase (MAPKKK).
To date, the MAPK pathways are mainly composed of extracellular signal-regulated kinase (ERK), Jun N-terminal kinase (JNK), and p38 MAPK signal pathways [14]. The ERK pathway is the best studied of the mammalian MAPK pathways. In the ERK pathway, ERK (ERK1 and ERK2) is activated upon phosphorylation by MEK (MEK1 and MEK2), which is itself activated by Raf (Raf-1, B-Raf, and A-Raf). The ERK pathway is activated by numerous extracellular signals and responsible for the activation of a number of transcription factors participating in cell growth, proliferation, and differentiation [7, 21]. PD98059 [19] is an effective ERK pathway-specific inhibitor. The c-Jun N-terminal kinase (JNK) cascade represents the other MAPK signaling pathway that is activated primarily by cytokines and exposure to environmental stress [6, 23]. The general substrate of JNKs is c-Jun but can also activate the transcription factors ATF-2, Elk-1, MEF-2c, p53, c-Myc and other factors such as Bcl-2, Bcl-xL [24, 25]. The JNK signaling pathway is mainly responsible for cell proliferation, apoptosis, and survival under stress and SP600125 is a JNK signaling pathway-specific inhibitor [15]. The cellular p38 cascade is another MAPK signaling pathway and contributes to cell apoptosis and cell cycle regulation. The mammalian p38 pathway can be activated by a wide range of cellular stresses, such as inflammatory cytokines, pathogens, osmotic pressure, heat shock, ultraviolet radiation, oxidative stress, and mitogens, and can also be specifically inhibited by the p38 pathway inhibitor, SB203580 [5].
Previous studies have shown that HIV-1 infection can activate the host cellular MAPK signaling pathway by the viral proteins, gp120 [9, 18], Tat [20], and Nef [16]. Conversely, activation of the MAPK signaling pathway can enhance HIV-1 gene expression level, viral genome replication level, and virus infection activity [3, 11, 26]. These studies have provided a new insight in the prevention and control of HIV-1 infection. In the current study, we further studied the contribution of each MAPK signal pathway com-ponent among the ERK, JNK, and p38 pathways to HIV-1 infection using pathway-specific inhibitors alone and in combination.
HTML
-
Human embryonic kidney fibroblast (293T) and human epithelial carcinoma cell lines (HeLa) were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, and 100 mg/mL streptomycin. The human T-lymphocyte cell line, MT-2, was maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum, 100 IU/mL penicillin, and 100mg/mL streptomycin. MAPK pathway inhibitors PD98059 (S805), SB203580 (S1863), and SP600125 (S1876) were purchased from Beyotime (Hangzhou, China); HIV-1 p24 Antigen ELISA kit (0801111) was purchased from ZeptoMetrix (Franklin, USA). Lipofectamine 2000, Platinum SYBR Green QPCR SuperMix-UDG with ROX, and Trizol reagent were purchased from Invitrogen (Carlsbad, USA). DNeasy Blood & Tissue Kit and RNeasy Mini Kit were purchased from Qiagen (Valencia, USA). Dual-Luciferase Reporter Assay System and Luciferase Assay System were purchased from Promega Corporation (Madison, USA).
-
The pNL4-3 plasmid contains an infectious HIV-1 provirus [1]. The plasmid, pNL4-3-LucR-E-, is a pNL4-3 clone that contains an Env-inactivating mutation and a firefly luciferase gene replacing nef [4]. The pGL3-LTR was constructed by PCR amplifi-cation of the long terminal repeat (LTR) region from pNL4-3 and subsequently cloned into pGL3-basic vector. The pAP-1-luc, p53-luc, pNF-κB-luc, and pSRE-luc were inducible reporter plasmids containing the Photinus pyralis (firefly) luciferase reporter gene driven by a basic promoter plus the corresponding cis-enhancer element. The pRL-TK plasmid (Promega) was used as a control plasmid that expresses Renilla luciferase reporter gene driven by the herpes simplex virus thymidine kinase promoter, and was used to monitor the efficiency of transfection. The pAP-1-luc and p53-luc were provided by Dr. Lin-ding Wang (Wuhan Institute of Virology, CAS, China). The pVpack-VSV-G plasmid was provided by Dr. Zheng-li Shi (Wuhan Institute of Virology, CAS, China).
-
Replication component NL4-3 virus stock was prepared by harvesting the supernatant of 293T cells transfected with pNL4-3 plasmid. VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus was produced by co-transfecting 293T cells with the pNL4-3-LucR-E-and pVPack-VSV-G plasmids. The infectious titer of the HIV-1 NL4-3 virus stock was measured by end-point dilution in 96-well cell culture plates on MT2 cells. The VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus stock was quantitated by p24 ELISA according to the manufacturer's directions. For VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus infection, HeLa cells were seeded in 24-well cell culture plates and infected with the same amount of VSV-G pseudotyped HIV-1NL4-3luciferase reporter viruses. After 4 h, infected HeLa cells were treated with MAPK signaling pathway inhibitors. The working concentration of each MAPK signaling pathway inhibitor was referred to the manufacturer's directions and previous reporters. Luciferase activity was measured after 48 h with the Luciferase Assay System (Promega) according to the manufacturer's directions. For the HIV-1NL4-3 infections, the MT2 cells were seeded in 24-well cell culture plates and infected with the same amount of HIV-1NL4-3 virus. After 4 h, infected HeLa cells were treated with MAPK signaling pathway inhibitors. Cell-free supernatants of infected HeLa cells were collected, and p24 concentration and RT activity were detected at 24, 36, 48, 60, 72, 84 and 96 h post-infection according to the manufacturer's protocol.
-
HeLa cells (~1×105) were seeded on 12-well dishes and transfected with pAP-1-luc, p53-luc, pNF-κB-luc, pSRE-luc, and pGL3-LTR the following day using Lipofectamine 2000. To normalize for transfection efficiency, the thymidine kinase (TK)-Renilla luciferase reporter plasmid (pRL-TK) was added to each transfection. At 4 h post-transfection, cells were treated with MAPK signaling pathway inhibitors, and 48 h post-transfection, dual-specific luciferase reporter gene assays was performed using Dual-Luciferase Reporter Assay System according to the manu-facturer's directions. Firefly luciferase activities were normalized based on the activities of Renilla luciferase.
Cell culture and reagents
Plasmids and constructs
Viruses and infections
Luciferase reporter gene assay
-
Previous studies have shown that HIV-1 infection efficiency is dependent on MAP kinase activation [11]. To further elucidate the contribution of each MAPK signal pathway to HIV-1 infection, we first investigated the effect of each MAPK pathway inhibitor and of their combination on VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus infection. As shown in Fig. 1, when treated the VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus infected HeLa cells separately with 50µmol/L PD98059, 10µmol/L SB203580 or 10µmol/L SP600125, PD98059 produced the highest VSV-G pseudotyped HIV-1NL4-3 inhibition activity, SB203580 and SP600125 also produced marked VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus inhibition activity compared with the DMSO control. When cells were treated with a combination of the three pathway inhibitors, the VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus inhibition activity increased significantly compared with each inhibitor alone.

Figure 1. Effects of MAPK pathway inhibitors on VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus infection. HeLa cells were seeded in 12-well cell culture plate and infected with HIV-1NL4-3 luciferase reporter virus the following day. Four hours after infection, cells were treated with 50µmol/L PD98059 (PD), 10µmol/L SB203580 (SB), 10µmol/L SP600125 (SP), 50µmol/L PD98059 plus 10µmol/L SB203580 (PD+SB), 50µmol/L PD98059 plus 10µmol/L SB203580 and 10µmol/L SP600125 (PD+SB+SP), or DMSO (negative control), respectively. At 48 h post-infection, cells were lysed for luciferase detection. All values represent the means and SD of three assays.
-
To further validate the HIV-1 inhibition activity by MAPK pathway inhibitors, we studied the effects of the three MAPK pathway inhibitors on HIV-1NL4-3 virus infection. We treated HIV-1NL4-3 virus infected MT2 cells with 50µmol/L PD98059, 10µmol/L SB203580, 10µmol/L SP600125 separately or in combination, and detected the p24 concentration and RT activity of the infected cells. As shown in Fig. 2A, when the HIV-1NL4-3 virus infected MT2 cells were treated with 10µmol/L SB203580 or 10µmol/L SP600125 separately, there was no effect on HIV-1NL4-3 p24 production compared with DMSO control. However, treatment of cells with 50µmol/L PD98059 separately or 50µmol/L PD98059 plus 10µmol/L SB203580 can reduce HIV-1NL4-3 p24 production significantly compared with the DMSO control. Additionally, 50µmol/L PD98059 plus 10µmol/L SB203580 and 10µmol/L SP600125 induced the highest HIV-1NL4-3 p24 inhibition activity. Similarly, as indicated in Fig. 2B, 10µmol/L SB203580 or 10µmol/L SP600125 had no effect on HIV-1NL4-3 RT activity compared with DMSO control when used separately. Treatment of cells with 50µmol/L PD98059 separately or 50µmol/L PD98059 plus 10µmol/L SB203580 inhibited HIV-1NL4-3 RT activity significantly compared with the DMSO control. Moreover, 50µmol/L PD98059 plus 10µmol/L SB203580 and 10µmol/L SP600125 have the highest HIV-1NL4-3 RT activity inhibition ability, and can completely restrict HIV-1NL4-3 RT activity of the infected cells.
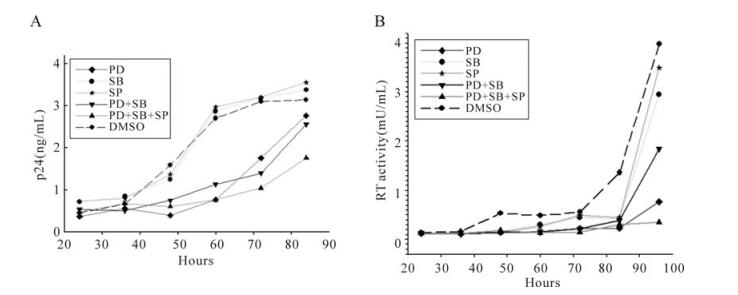
Figure 2. Effects of MAPK pathway inhibitors on HIV-1NL4-3 virus infection. MT2 cells were seeded in 12-well cell culture plate and infected with HIV-1NL4-3 at an MOI of 1. At 4 hours post-infection, culture medium was substituted and the MT2 cells were treated with 50µmol/L PD98059 (PD), 10µmol/L SB203580 (SB), 10µmol/L SP600125 (SP), 50µmol/L PD98059 plus 10µmol/L SB203580 (PD+SB), 50µmol/L PD98059 plus 10µmol/L SB203580 and 10µmol/L SP600125 (PD+SB+SP), or DMSO (negative control) respectively. At 24, 36, 48, 60, 72, 84 and 96 h post-infection, cell-free supernatants of the infected cells were collected for p24 detection (A) and reverse transcriptase activity detection (B).
-
The data above have shown that the ERK pathway inhibitor, PD98059, can restrict HIV-1NL4-3 virus infection and VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus infection, and the p38 MAPK pathway inhibitor, SB203580, and JNK pathway inhibitor, SP600125, can enhance the inhibition efficiency. To further investigate the effects of MAPK pathway inhibitors on HIV-1 replication, we studied the effects of the MAPK inhibitors on HIV-1 LTR promoter activity. We cloned HIV-1NL4-3 LTR promoter in upstream of the firefly luciferase reporter gene in pGL3-basic (Promega), co-transfected HeLa cells with pGL3-LTR and Renilla luciferase vector (pRL-TK) and the tran-sfected cells treated with 50µmol/L PD98059, 10µmol/L SB203580, 10µmol/L SP600125 separately or in combination. As a result, the relative luciferase activity of the HIV-1 LTR promoter was potently inhibited by the ERK pathway inhibitor, PD98059. However, the p38 MAPK pathway inhibitor, SB203580, and the JNK pathway inhibitor, SP600125, can only slightly restrict HIV-1 LTR promoter activity, and SB203580 and SP600125 have no effects on the HIV-1 LTR promoter inhibition activity by PD98059 (Fig. 3).
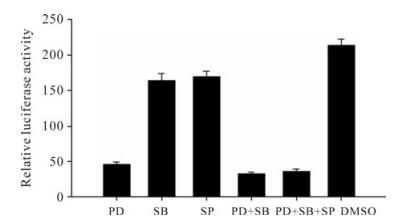
Figure 3. Effects of MAPK pathway inhibitors on HIV-1 LTR promoter activity. HeLa cells were seeded in 12-well cell culture plate and co-transfected with 0.4 µg pGL3-LTR and 0.04 µg pRL-TK. At 4 hours post-infection, cells were treated with 50µmol/L PD98059 (PD), 10µmol/L SB203580 (SB), 10µmol/L SP600125 (SP), 50µmol/L PD98059 plus 10µmol/L SB203580 (PD+SB), 50µmol/L PD98059 plus 10µmol/L SB203580 and 10µmol/L SP600125 (PD+SB+SP), or DMSO (negative control), respectively. At 48 h post-infection, cells were lysed for luciferase detection. All values represent the means and SD of three assays.
-
As previously reported, the HIV-1 LTR promoter activity is dependent on a wide range of cellular transcription factors, including NF-κB, AP-1, p53 and SRE [17]. It was also reported that NF-κB, AP-1, p53 and SRE were involved in the MAPK signaling pathway and responsible for the regulation of MAPK pathway-related genes [8, 13]. The above data show that HIV-1 LTR promoter activity was down-regulated by MAPK pathway inhibitors. To further validate the direct relationship between MAPK pathway inhibitor and the down-regulation of HIV-1 LTR promoter activity, we investigated the effects of MAPK pathway inhibitors on transcription factors by transfecting HeLa cells with pAP-1-luc, pSRE-luc, pNF-KB-luc or p53-luc reporter plasmids and treated the transfected cells with MAPK inhibitors. The results indicated that 50µmol/L PD98059 had strong negative effects on pAP-1-luc and pNF-KB-luc reporter plasmids (Fig. 4A, C), and only slight effects on pSRE-luc and p53-luc reporter plasmids (Fig. 4B, D). Additionally, we found that 10µmol/L SB203580 and 10µmol/L SP600125 had no obvious effects on the four reporter plasmids when used separately. When used in combination, 10µmol/L SB203580 and 10µmol/L SP600125 significantly enhanced the inhibition activity of 50µmol/L PD98059 on the pSRE-luc reporter plasmid (Fig. 4B).
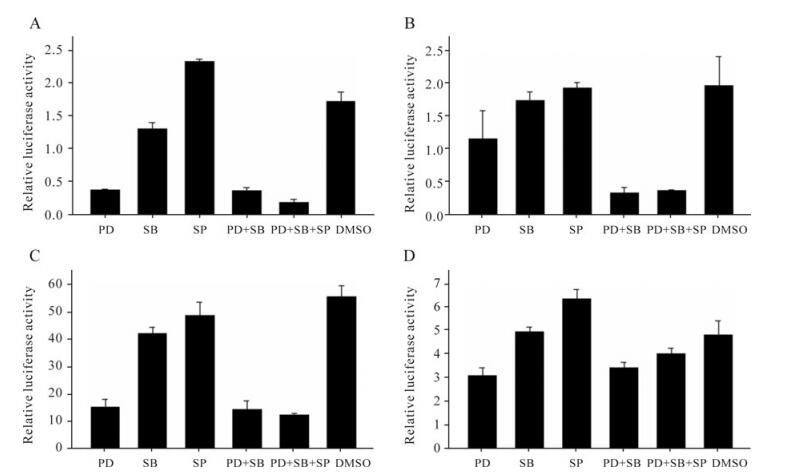
Figure 4. Regulation of transcription factors by MAPK pathway inhibitors. HeLa cells were seeded in 12-well cell culture plate and co-transfected with 0.6 µg pAP-1-luc and 0.006 µg pRL-TK(A), 0.6 µg pSRE-luc and 0.04 µg pRL-TK(B), 0.6 µg pNF-KB-luc and 0.06 µg pRL-TK(C), and 0.6 µg p53-luc and 0.04 µg pRL-TK(D), respectively. At 4 hours post-infection, cells were treated with 50µmol/L PD98059 (PD), 10µmol/L SB203580 (SB), 10µmol/L SP600125 (SP), 50µmol/L PD98059 plus 10µmol/L SB203580 (PD+SB), 50µmol/L PD98059 plus 10µmol/L SB203580 and 10µmol/L SP600125 (PD+SB+SP), or DMSO (negative control) respectively. At 48 h post-transfection, cells were lysed for luciferase detection. All values represent the means and SD of three assays.
Inhibition of VSV-G pseudotyped HIV-1NL4-3 luciferase reporter virus infection by MAPK pathway inhibitors
Inhibition of HIV-1NL4-3 virus infection by MAPK pathway inhibitors
Effects of MAPK pathway inhibitors on HIV-1 LTR promoter activation
Down-regulation of transcription factors by MAPK pathway inhibitors
-
Previous studies have shown that HIV-1 infection can activate the host cellular MAPK signaling pathway, and again, conversely, an activated MAPK signal path-way can enhance HIV-1 infection efficiency [11, 12, 18, 26]. In this study, we took advantage of three available MAPK signal pathway inhibitors, the ERK pathway inhibitor, PD98059, the JNK pathway inhibitor, SP600125, and the p38 pathway inhibitor, SB203580, and further investigated the contribution of each MAPK signaling pathway to HIV-1 virus replication. We found that 50µmol/L PD98059 can significantly restrict HIV-1 infection, 10µmol/L SB203580 and 10µmol/L SP600125 only have slight HIV-1 inhibition activity but can enhance the HIV-1 inhibition activity of PD98059 when used in combination. This finding could be explained by the possibility that the ERK pathway is responsible for the main contribution to MAPK signal pathways for HIV-1 virus infection and replication, whereas JNK and p38 pathways only weakly regulate HIV-1 infection and replication. Therefore, when the ERK pathway was blocked, HIV-1 infection was significantly restricted. However, the MAPK signaling pathway can also enhance HIV-1 infection efficiency to some extent through the JNK and p38 pathways. When the JNK or p38 pathways were blocked, HIV-1 infection was slightly decreased because HIV-1 virus can infect and replicate freely through the ERK pathway. However, when all three MAPK signal pathways were blocked, HIV-1 infection and replication was restricted to the maximum extent.
Previous studies have also shown that activation of the MAPK signaling pathway can enhance HIV-1 LTR promoter activity [2, 27]. Therefore, the current study investigated whether the inhibition of HIV-1 infection was a result of inhibition of HIV-1 LTR promoter activity. We found that the ERK pathway inhibitor, PD98059, significantly reduced HIV-1 LTR promoter activity, whereas the JNK pathway inhibitor, SP600125, and the p38 pathway inhibitor SB203580, only slightly reduced HIV-1 LTR promoter activity. These data are consistent with the above data for MAPK pathway inhibitors on HIV-1 infection. Furthermore, we found that the transcription factor reporter plasmids corresponding to HIV-1 LTR promoter were also strongly restricted by the ERK pathway inhibitor, PD98059, and only slightly restricted by the JNK pathway inhibitor, P600125, and p38 pathway inhibitor, SB203580. These data are also consistent with the above data for MAPK pathway inhibitors on HIV-1 infection.
In conclusion, our data indicate that the MAPK inhibitors restrict HIV-1 infection through reducing HIV-1 LTR promoter activity by inhibiting the transcription factors corresponding to HIV-1 LTR promoter. The ERK pathway inhibitor, PD98059, contributed the most to HIV-1 infection and HIV-1 LTR promoter activity inhibition of the three distinct MAPK signaling pathway inhibitors. The JNK pathway inhibitor, P600125, and the p38 pathway inhibitor, SB203580, had only slight effects on HIV-1 infection and HIV-1 LTR promoter activity but can enhance the inhibition efficiency of the ERK pathway inhibitor, PD98059.

 DownLoad:
DownLoad: