












HTML
-
Coxsackievirus A16(CA16), a non-enveloped, single positive-stranded RNA virus, is a member of the genus Enterovirus, which belongs to the Picornaviridae family(Poyry et al., 1994; Mao et al., 2012; Li et al., 2013).The viruses in this family possess a homology of 64% and 72% at nucleotide and amino acid levels, respectively(Oberste et al., 1999). The length of the CA16 genome is approximately 7, 410 nucleotides, consisting of a single long open reading frame(ORF), flanked by the 5'-and 3'-untranslated regions(UTRs). The ORF encodes a large polyprotein precursor, which can be further cleaved into structural protein P1 that encodes four structural capsid proteins(VP1, VP2, VP3, and VP4), and nonstructural proteins P2 and P3 that encode seven non-structural proteins(2A, 2B, 2C, 3A, 3B, 3C and 3D)responsible for viral replication and protein processing(Puenpa et al., 2011; Han et al., 2014; Xu et al., 2013; Wei et al., 2014). The four capsid proteins form the icosahedral virion structure, with VP1, VP2, and VP3 exposed on the surface, and VP4 arranged internally(Shi et al., 2013). Among these capsid proteins, the major antigenic and neutralization epitopes mainly reside on VP1(Lim et al., 2013), which therefore plays a critical role in the process of infection(Zhang et al., 2012). Based on their VP1 sequences, CA16 strains can be classified into two genogroups, A and B, with the latter consisting of two subgenotypes B1(B1a, B1b, and possibly B1c) and B2(Perera et al., 2007; Zhang et al., 2010; Zhou et al., 2011).
CA16 is one of the major pathogens of human hand, foot, and mouth disease(HFMD)(Iwai et al., 2009; Cai et al., 2013), occurring mainly in young children(Zhang et al., 2012; Han et al., 2014). Individuals infected with CA16-associated HFMD usually show mild symptoms(Man-Li et al., 2012), such as fever, oral ulcers, and rashes on the surface of hands, feet, and buttocks. However, many recent reports demonstrate that CA16 infections may lead to severe complications, including aseptic meningitis, rhombencephalitis, cardiac and pericardial disease, pulmonary complications, and spontaneous abortion, ultimately leading to lethal myocarditis and pneumonia(Mao et al., 2012; Mao et al., 2013; Shi et al., 2013). Co-circulation of CA16 and EV71 has been described in HFMD outbreaks in China, Malaysia, and Taiwan(Han et al., 2014), which makes the control and management of HFMD epidemic more complex and difficult. In addition, the coinfection of CA16 and EV71 often results in serious complications of the central nervous system(CNS), with longer duration and higher rates of critical illness transmission. During the course of coinfection, the risk of gene recombination within these RNA viruses may increase(Mao et al., 2012). A recent clinical survey indicated that 38.3% of 266 laboratory-confirmed HFMD cases were caused by CA16 infection, during the HFMD outbreak in China in 2009(Shi et al., 2013). CA16 infection poses a severe potential health threat to children, necessitating the need for investigation of replication mechanisms of, and vaccine development for, the CA16 virus.
We constructed an infectious cDNA clone of an epidemic strain of CA16, and developed an eGFP reporter virus, following a previously described strategy for the construction of an infectious cDNA clone of EV71(Shang et al., 2013). Upon transfection of RNA, transcribed from the full-length cDNA clone, into Vero cells, the expression of viral proteins, production of progeny viruses, and specific infectivity collectively suggested that the recombinant virus is highly infectious. The recombinant and parental viruses showed similar biological properties in terms of plaque morphology and growth kinetics. Furthermore, the eGFP reporter virus enables real-time monitoring of viral replication and spread in infected cells, without the need for traditional, time-consuming plaque assays and antibody-specific detection methods. The eGFP-CA16 reporter virus provides a powerful tool for monitoring and visualizing dynamic processes of viruses in living cells, and performing high-content screening(HCS)for the identification of potential inhibitors of CA16.
-
Vero(African green monkey kidney)cells used in this study were maintained in Dulbecco's modified Eagle's medium(DMEM)supplemented with 10% FBS, 100 U/ mL of penicillin, and 100 μg/mL of streptomycin. Cells were incubated at 37 ℃ with 5% CO2. The parental strain CA16/GD09/24 and the mouse polyclonal antibody against CA16 VLP were provided by Cheng-Feng Qin's lab in the State Key Laboratory of Pathogen and Biosecurity, Beijing Institute of Microbiology and Epidemiology.
-
Viral RNA was extracted from the parental virus using QIAamp Viral RNA Mini Kit(Qiagen, Germany). The first strand of cDNA, containing the full-length CA16 genome, was synthesized by reverse transcription using a High Fidelity PrimeScript RT-PCR Kit(Takara, Japan)with oligo(dT)primers. The cDNA was used as a template for constructing the full-length cDNA clone of Coxsackievirus A16, according to the cloning strategy shown in Figure 1.
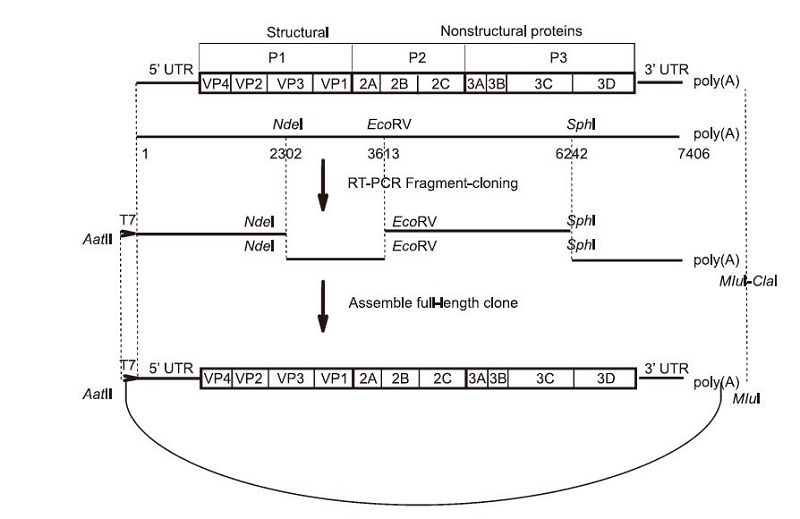
Figure 1. Schematic representation of the full-length cDNA clone of CA16. Four primer pairs were designed based on a template sequence from GenBank accession No KC117317. Four cDNA fragments: fragment (T7+1–2441), fragment (2101–3782), fragment (3511–6362), and fragment (5940–7409+poly(A)27), represented by thick lines, were amplified by the primer pairs from the reverse-transcribed first strand of cDNA, to cover the complete CA16 genome. The unique restriction sites used in the assembly of the full-length cDNA clone of CA16 (pACYC-CA16-FL), and their nucleotide numbers, are shown in the schematic representation
-
The full-length cDNA clone pACYC-CA16 was constructed according to the following procedure: four fragments covering the complete genome were amplified from the reverse transcribed first strand cDNA. Fragment(T7+1-2441) was amplified using primer pairs T7-CA16-5'-UTR-F/CA16-2441-Sma Ⅰ-R. Similarly, fragment(2101-3782), fragment(3511-6362), and fragment(5940-7409+poly(A)27)were amplified using primer pairs CA16-2101-Aat Ⅱ-F/CA16-3782-Sma Ⅰ-R, CA16-3511-Aat Ⅱ-F/CA16-6362-Sma Ⅰ-R, and CA16-5940-Aat Ⅱ-F/CA16-poly(T)-Mlu Ⅰ-Cla Ⅰ, respectively. Next, the four fragments were cloned and inserted into pACYC177 between the restriction sites Aat Ⅱ and Sma Ⅰ/Cla Ⅰ, generating CA16-A, CA16-B, CA16-C, and CA16-D. Then, the subclone CA16-B was digested by Nde Ⅰ/Sma Ⅰ, and the fragment between Nde Ⅰ and Sma Ⅰ was ligated into Nde Ⅰ/Sma Ⅰ digested CA16-A to create CA16-AB. The subclone CA16-C was digested by Aat Ⅱ/Sph Ⅰ, and the fragment between Aat Ⅱ and Sph Ⅰ was ligated into the Aat Ⅱ-Sph Ⅰ sites of CA16-D, to construct plasmid designated as CA16-CD. Finally, the fragment between Aat Ⅱ and Eco RV from CA16-AB was inserted into CA16-CD using the Aat Ⅱ and Eco RV sites, thus yielding the fulllength cDNA clone pACYC-CA16.
The eGFP-CA16 reporter plasmid was constructed in multiple steps. The Bsi WI and Not Ⅰ sites, and 2A protease cleavage site(ITTLG)were introduced to pACYC-CA16 between the 5'-UTR and the N-terminus of VP4 via overlap PCR. Overlap PCR was performed using the products of the first two PCRs, and primer T7-CA16-5'-UTR-F/CA16-1586-R. The eGFP gene was simultaneously amplified using primers Bsi WI-eGFP-F and Not Ⅰ-eGFP-R from pEGFP-N1(Clontech, Japan), and then inserted between the Bsi WI and Not Ⅰ sites to generate pACYC-eGFP-CA16.
All primers used for PCRs are listed in Table 1. All constructs were confirmed by sequencing.

Table 1. Primer sequences used in this study
-
Recombinant CA16 and eGFP-CA16 RNAs were transcribed from corresponding Mlu Ⅰ-linearized cDNAs with the MEGAscript kit(Ambion, USA), as recommended by the manufacturer. After treatment with DNase Ⅰ(for 15 min at 37 ℃), LiCl was added to the transcription mixture, and precipitated RNA was washed with 70% ethanol, followed by quantification by spectrophotometry. The transcripts were analyzed on an agarose gel after electrophoresis.
In vitro transcribed RNAs, from pACYC-CA16 and pACYC-eGFP-CA16, were transfected into Vero cells using DMRIE-C reagent(Invitrogen, USA). Transfection was performed in 35 mm dish with 80% confluent monolayers of Vero cells. 4 μL of DMRIE-C was added into 1 mL Opti-MEM Medium(Invitrogen), with brief agitation, and 1 μg of the RNA transcripts were then added into the mixture immediately. The diluted RNA-DMRIE-C complex solution was then added directly to the dish of Vero cells, and incubated at 37 ℃ in a 5% CO2 incubator. On observation of cytopathic effect(CPE), the supernatant containing rescued viruses was harvested and stored at −80 ℃ for later use.
-
Vero cells were seeded in 24-well plates(Corning)with a density of 1.0×105 cells/well, at 37 ℃ overnight. The Vero cell monolayers were infected with ten-fold serial dilutions of virus-containing culture fluids, at a volume of 100 μL per well. After 1 h of incubation at 37 ℃ with 5% CO2, the culture was discarded, and medium mixed with 2% methyl cellulose was added to each well. After 3 days post-infection, the layer was discarded, and cells were fixed with 3.7% formaldehyde and stained with 1% crystal violet. The plaque-forming units(PFU)per mL were calculated.
-
Vero cells transfected with in vitro-transcribed fulllength CA16 RNA were seeded on glass coverslips. Cells were washed with PBS and fixed by cold(−20 ℃)5% acetic acid in acetone for 15 min at room temperature 24, 36, and 48 h post-transfection(hpt). After washing three times with PBS, cells were incubated with mouse polyclonal antibody against CA16 VLP(1: 250 dilution with PBS)for at least 1 h. Subsequently, the cells were incubated with goat anti-mouse IgG conjugated with Texas-Red, at a 1:125 dilution with PBS, and then at room temperature for another hour after three washes with PBS. Finally, cell nuclei were stained with 4, 6-diamidino-2-phenylindole(DAPI)for approximately 5 min. The fluorescence signal was analyzed using a NIKON upright fluorescence microscope.
-
RNA was extracted from CA16-infected Vero cells using Trizol reagent(TaKaRa). The extracted RNA was identified by RT-PCR using PrimeScript One Step RTPCR Kit(TaKaRa), with specific primer CA16-2374-F/ CA16-3039-R targeted to the VP3-VP1 genes.
-
Vero cells were seeded in 12-well plates, at a density of 1.0×105 cells/well. Cells were inoculated with eGFP-CA16 virus, at a multiplicity of infection(MOI)of 0.1 at 37 ℃ with 5% CO2. After 1 h, the culture was discarded, and fresh maintained DMEM containing different concentrations of NITD008 was added to each wells. Supernatants were collected after incubation at 37 ℃ for 48 h. The viral titers were quantified by plaque assay. The 50% effective concentration(EC50)value of NITD008 was calculated by the Behrens-Kärber method(Zhang et al., 2015).
Cell culture, viruses, and antibodies
Viral RNA extraction and cDNA procedure
Construction of plasmids
In vitro RNA transcription and transfection
Plaque assay
Immunofluorescence assay(IFA)
RNA isolation and RT-PCR
Viral inhibition assay
-
To construct the full-length CA16 cDNA clone, viral RNA was extracted and reverse transcription was performed. Four fragments were amplified by reverse transcription, and cloned into pACYC177. The four subclones were assembled into the full-length CA16 cDNA clone as shown in Figure 1. The full-length plasmid was linearized by Mlu Ⅰ. A T7 promoter sequence was engineered upstream of the 5'-UTR, and the linearized plasmid thus obtained could be used as a template for in vitro transcription. A genome-like RNA molecule with a 5' non-viral G nucleotide derived from the T7 promoter, and a poly(A)tail at its 3' terminus, was generated.
With the aim of constructing a reporter virus, a CA16 genome carrying the eGFP gene was constructed. Reporter genes were inserted between the 5'-UTR and the N-terminus of VP4, with the addition of a CA16 2A protease cleavage site(ITTLG)at its C-terminus(Figure 3A).
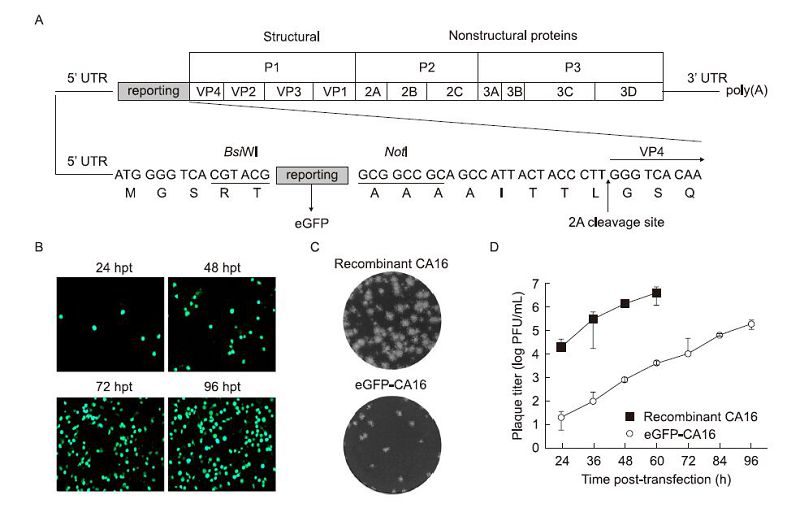
Figure 3. Diagram of the cloning procedure used for introducing eGFP into the CA16 genome, and characterization of the eGFP-CA16 reporter virus. (A) Construction of the eGFP-CA16 reporter virus. The eGFP reporter gene and the Bsi WI-NotI enzyme sites and 2A protease cleavage site (ITTLG) were introduced into pACYC-CA16 for expression of eGFP. (B) Expression of eGFP in Vero cells. The eGFP-CA16 reporter RNA transcripts were analyzed by fluorescence microscopy at different times post-transfection. Plaque phenotypes (C), and kinetics of recombinant CA16 and eGFP-CA16 viruses (D). Three independent experiments were performed in duplicate, with error bars representing standard deviation.
-
Recombinant CA16 RNA was transcribed from the Mlu Ⅰ-linearized full-length cDNA plasmid by T7 RNA polymerase. Approximately 50 to 70 μg of RNA were mixture. In total, 2.5 × 105 Vero cells were transfected with 1 μg of RNA transcripts, as described in the Materials and Methods section. Apparent CPE was observed in cells on day 2 post-transfection(data not shown). To assessing the production of viruses, IFA was used to detect viral protein expression in transfected cells. As shown in Figure 2A, the number of cells expressing viral protein increased from 24 hpt to 48 hpt, and more than 60% cells were IFA-positive at 48 hpt. The culture medium from the transfected cells was collected at different time points, and virus titers were determined in Vero cells by plaque assays. Increasing amounts of viral titer were observed, and the highest viral titers(1.6 × 107 PFU/mL)were obtained at 60 hpt(Figure 2B). The recombinant virus showed no difference from the parental virus, in terms of plaque size or morphology in Vero cells(Figure 2C). Furthermore, we observed similar viral growth ki-netics at MOI of 0.1 for the parental and recombinant viruses(Figure 2E). Virus-specific RT-PCR was performed to confirm the release of virions for recombinant viruses. The recombinant and parental viruses produced the same size amplicons in virus-specific RT-PCR assay(Figure 2D). Our results showed that genome-length CA16 RNA could be transcribed in vitro from the constructed cDNA clone, and that the recombinant virus exhibited similar characteristics to the parent virus.

Figure 2. Production and characterization of the recombinant infectious CA16 virus. (A) The level of viral protein expression analyzed by IFA. The full-length CA16 RNA transcript was transfected into Vero cells and viral protein expression was analyzed by IFA, at the indicated times post-transfection. (B) Supernatants were collected at different times post-transfection, and viral titers were calculated as the number of plaque-forming units (PFU) per microliter, by plaque assays. (C) Viral plaque phenotypes of parental and recombinant CA16 in Vero cells. (D) RTPCR identification of the rescued virus. RNA extracted from transfected or infected cells was reversely transcribed, followed by PCR using primers specific to viral genes. Lane 1: Marker Ⅲ; Lane 2: PCR product of RNAtransfected cells; Lane 3: PCR product of parental virus infected cells; Lane 4: PCR product of mock-transfected cells. (E) Growth kinetics of parental and recombinant CA16 virus. Vero cell monolayers were infected with viral supernatants at MOI = 0.1, and the virus yield was determined at the indicated time points by plaque assay. Results are expressed as the average of three independent experiments, and representative data are presented.
As described above, eGFP-CA16 reporter virus was also produced by transfecting Vero cells with the corresponding RNA transcripts. The eGFP signal representing the level of protein expression was monitored by fluorescence microscopy, at different times post-transfection. The percentage of eGFP positive cells among the infected cells increased with time(Figure 3B), and this increase was accompanied by a CPE resulting in > 90% cell death, at 4 days post-transfection. The growth kinetics of the reporter virus were compared to those of the recombinant CA16 at different time points after transfection, and analysis of plaque morphology of reporter and recombinant viruses was performed by plaque assay. Our results showed that eGFP-CA16 showed smaller plaque size than the recombinant full-length CA16 virus(Figure 3C). Although the viral titer of eGFP-CA16 was approximately 1000-fold lower than that of recombinant CA16, both viruses displayed similar growth trends(Figure 3D). Because the recombinant CA16 RNA transfected cells were all dead after 60h post-transfection, therefore no more supernatants were harvested at 72h and 96h postinfection.
-
The adenosine nucleoside analog NITD008, which is a selective inhibitor used to target the family Flaviviridae, has been reported to potently inhibit wild-type CA16(CA16-WT)in cell culture at a 50% effective concentration(EC50)value of 0.64 μmol/L(Deng et al., 2014). In order to confirm the efficacy of NITD008 inhibition of eGFP-CA16 replication, we investigated the effects exerted by this compound, using Vero cells infected with eGFP-CA16 at an MOI of 0.1, at different concentrations. eGFP expression in infected cells occurred in a dose-dependent manner(Figure 4A), consistent with the variation observed in virus titers derived from supernatants collected at corresponding concentrations of NITD008(Figure 4B). The EC50 of NITD008 for eGFP-CA16 was found to be 0.63 μmol/L, which was similar to that observed for CA16-WT at the same concentration. It is suggested that the eGFP-CA16 reporter virus possesses similar properties to CA16-WT in terms of antiviral activity, and may be used in studies of potential antiviral agents against CA16.
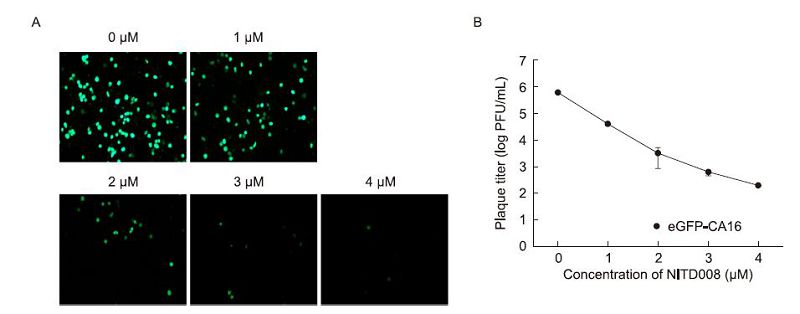
Figure 4. Correlation between eGFP expression levels and CA16 replication, when NITD008 is added. (A) Vero cells were infected with eGFP-CA16 at MOI of 0.1, and different concentrations of NITD008 were added. After 48h, the eGFP expressions were detected by fluorescence microscopy. (B) Viral titer reduction assays were used to assess the antiviral activity of NITD008 against eGFP-CA16 replication. Data were from three independent experiments with error bars representing SD from the mean
Construction of the recombinant CA16 virus cDNA clone and the genome-length reporter virus
Generation and characterization of recombinant CA16 and eGFP-CA16 viruses
Assessment of the inhibitory effect of NITD008 on eGFP-CA16 replication
-
Recent epidemiological surveys have indicated that CA16 has become more virulent in humans, causing a large number of deaths and severe cases of neurological complications(Wang et al., 2004; Goto et al., 2009). No approved vaccines or efficient antivirals are available for CA16 infection(Shi et al., 2013). It is vital to focus research efforts on the development of a validated and rapid screening assay system in the search for potential antiviral compounds. In this study, a recombinant infectious cDNA clone of CA16 was constructed, based on the reverse-genetic system. The results of IFAs demonstrate that the infectious CA16 virus can be rescued by transfection of in vitro transcribed CA16 RNA into Vero cells. Furthermore, in vitro growth kinetics and plaque assay also revealed that recombinant CA16 viruses exhibit similar proliferation rates and plaque morphology to the parental viruses. Our study provides a valuable genetic system to study CA16 infection and host interaction, which is expected to be essential for the development of vaccines and antivirals for the prevention and treatment of CA16 infections.
Many studies have indicated that viruses engineered to carry a fluorescent report gene may be readily used for high throughput antiviral screening assays, as the fluorescence signal generated is indicative of the antiviral activity of the inhibitor compounds screened.(Shang et al., 2013).
We constructed an eGFP-CA16 reporter virus cDNA clone by inserting the fluorescent reporter gene between the 5'-UTR and N-terminus of VP4, with the addition of a CA16 2A protease cleavage site(ITTLG)at its C-terminus. Although the viral titer for the reporter virus was observed to be lower, its growth kinetics were found to show a similar trend to those of the recombinant CA16 virus. In addition, the results of the antiviral activity assay suggest that the eGFP-CA16 reporter viruses have a comparable EC50 of NITD008 to CA16-WT, indicating that this reporter virus should be a powerful tool for screening potential antiviral agents of CA16.
In conclusion, we have successfully constructed a stable infectious cDNA clone that provides a valuable genetic system to study the molecular mechanisms of replication, infection, and pathogenesis in CA16. Furthermore, the eGFP-CA16 reporter virus is a very efficient and useful tool for screening antiviral agents in the discovery of drugs for the treatment and management of CA16 infections.
-
This work was supported by the Science and Technology Plan Projects of Wuhan(grant No. 2013060501010157). We thank the Center for Instrumental Analysis and Metrology, Wuhan Institute of Virology and Wuhan Key Laboratory on Emerging Infectious Diseases and Biosafety for helpful assistance during the course of the work.
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
BZ and XL designed the experiments. CD and LX carried out the experiments. HY, CQ and BZ analyzed the data. CD and SL wrote the paper. All authors read and approved the final manuscript.

 DownLoad:
DownLoad: