












HTML
-
Flaviviruses are enveloped, positive-stranded RNA viruses belonging to the Flaviviridae family (for a review on the biology of flaviviruses, consult (Best 2016)). The flavivirus genome encodes a single protein that is cut into three structural (capsid, C, membrane, M, and envelope, E) and seven nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) proteins (Guzman et al. 2010). These proteolytic cleavages are achieved by the viral protease NS2B/ NS3 and by cellular proteases, whereas the flaviviral RNA genome is replicated by the NS5 polymerase (Padmanabhan et al. 2006). Following clathrin-mediated endocytosis and entry into the cytoplasm by fusion, viral RNA replication occurs in replication complexes that are found within replication "organelles" on membranes of the endoplasmic reticulum (Aktepe and Mackenzie 2018). The formation of replication organelles is dependent on multiple viral nonstructural proteins as well as cellular proteins (Aktepe and Mackenzie 2018). Early translation of the incoming genome allows the synthesis of the nonstructural viral proteins necessary to genome replication, whereas late translation involves the genomes produced in the replication phase. Genome replication and translation occur in a tightly regulated fashion, along with assembly of novel flaviviral particles which also takes place at the endoplasmic reticulum (for a detailed review on flavivirus genome replication, consult (Mazeaud et al. 2018).
53 species of flaviviruses have been recognized, of which 40 can infect humans (Best 2006). Most flaviviruses that infect humans are zoonoses transmitted by arthropods and hence belong to the group of arboviruses (arthropod-borne) (Tabachnick 2016). Some are transmitted by ticks, such as the tick-borne encephalitis virus (TBEV) and Langat virus (LGTV), whereas others are transmitted by mosquitoes, mostly of the Aedes and Culex species, such as the Dengue virus (DENV), the Zika virus (ZIKV), the Japanese encephalitis virus (JEV), the West Nile virus (WNV) or the Yellow fever virus (YFV). The zoonotic reservoirs for flaviviruses include birds and non-human primates, but some flaviviruses (DENV, YFV, ZIKV) have also evolved to cycle between humans and arthropods (Best 2016). In addition, direct horizontal transmission among humans through sexual contacts is firmly demonstrated for ZIKV (D'Ortenzio et al. 2016) but has not been documented for other flaviviruses. The Asian lineage of ZIKV is also capable of vertical transmission by crossing the placental barrier (Brasil et al. 2016), a property that is also not seen in other flaviviruses. Flaviviral infections are often asymptomatic in humans, though many of them can induce disease and even death. Flavivirus-caused symptoms are relatively short and most often dependent on systemic inflammatory responses. DENV and YFV infect liver cells and may cause hemorrhagic fevers, whereas TBEV, ZIKV and WNV are neurotropic viruses that cause encephalitis (Gould and Solomon 2008). The Asian lineage of ZIKV is an outlier, as its vertical transmission causes gross fetal brain abnormalities, including microcephaly, that directly result from the virus destroying neural progenitors (Miner et al. 2016; Martinot et al. 2018). Innate and adaptive immune responses to flavivirus infections are highly efficient in humans, and often lead to long-term immunity (Augustine et al. 2010). Thus, flaviviruses have evolved to successfully infect two different host species, implying partial evasion from immune responses in both species. However, immune control in humans is generally efficient, and consequently flaviviruses rarely cause chronic infections. This review will focus on recent discoveries concerning type Ⅰ interferon (IFN-I)-stimulated, cell-autonomous innate responses to flavivirus infections in the mammalian host.
-
In its classical description, the cell-autonomous innate response to viral infections is based on the detection of viral molecules by specialized sensors, the pathogen recognition receptors (PRRs). PRRs activate signaling cascades leading to the transcriptional upregulation of hundreds of innate immunity-related factors, including the type Ⅰ interferon (IFN-I) cytokines which comprise IFN-α, IFN-β and other IFN subtypes. All IFN-I subtypes bind to a unique receptor, IFNAR (also called IFNα/βR) to activate the Janus kinase (Jak)/signal transduced and activator of transcription (STAT) secondary signaling cascade in an autocrine and paracrine manner, leading to the upregulation of dozens to hundreds of IFN-I-stimulated genes (ISGs) (Pestka et al. 2004; Takeuchi and Akira 2010; Liu et al. 2011). Among the well-described PRRs for flaviviruses are TLR3, TLR7, RIG-I and MDA5, which activate cellular innate immune pathways through the detection of viral genomic RNA structures (reviewed in (Diamond 2009; Wilkins and Gale 2010)). Activation of the IFN-I innate pathway has the potential to efficiently inhibit flavivirus replication, as shown for instance by multiple evidence of IFN-I treatment strongly inhibiting DENV infection in vitro (Diamond et al. 2000; Munoz-Jordan et al. 2003; Fink et al. 2007). In vivo, higher IFN-α plasma concentrations correlated with milder symptoms of the disease, suggesting an important role for these innate pathways in the control of DENV infection (De La Cruz Hernandez et al. 2014). Along these lines, patients with the more severe DENV infection (DHF patients with Dengue shock syndrome) express smaller levels of a number of ISGs compared to patients with the milder form of the disease (Simmons et al. 2007). In murine models, infection of IFNAR-knockout animals by WNV or ZIKV is highly pathogenic (Samuel and Diamond 2005; Yockey et al. 2016). Collectively, these findings support the idea that flaviviral infections may lead to an IFN-I response in vivo, and that this response has a crucial role in controlling the virus. Of note, cell-autonomous antiviral responses inhibiting flaviviruses do not always strictly require stimulation by IFN-I. The concept of "intrinsic immunity" refers to these responses that are immediate and rely on the pre-existing presence of basal levels of restriction factors in infected cells, hence not requiring cytokine-mediated signaling (Yan and Chen 2012). Also, induction of antiviral effectors can sometimes be achieved through IFN-I-independent pathways. For instance, the antiviral effector viperin was induced in Vero cells, which do not express IFN-I, following exposure to JEV, and was also induced in the presence of over-expressed JEV NS5, which blocks Jak-STAT signaling downstream of IFN-I binding to its receptor (Lin et al. 2006; Chan et al. 2008).
-
Upon infection of HaCaT keratinocyte cell monolayers by DENV followed by immunofluorescence microscopy analysis, expression of IFN-I-dependent antiviral factors seemed higher in non-productively infected bystander cells compared with the productively infected cells (Lopez-Gonzalez et al. 2018). Similar observations were made upon infection of primary monocyte-derived macrophages (MDMs) with DENV-2 (Helbig et al. 2013). In a murine model of WNV infection, uninfected bystander cells of the central nervous system similarly expressed elevated amounts of the antiviral protein viperin (Szretter et al. 2011). These observations strongly suggest that paracrine effects mediated by IFN-I and possibly by other molecules are important to the cellular protection against DENV, WNV and other flaviviruses. They also suggest that in the infected cells, the cell-intrinsic response may be inhibited by viral evasion activities (see below).
Paracrine Effect of IFN-I
-
The cell-autonomous response has inherently low specificity: engagement of PRRs will result in dozens or hundreds of gene products to be upregulated, including ISGs, but very few of theses proteins, if any, are effectors with the potential to directly inhibit infection by a given virus. Consequently, medium-to large-scale genetic screens have been performed in order to identify the restriction factors of flaviviruses. Two main approaches can be undertaken in such screens: gain-of-function, in which a library of overexpressed proteins is used in order to induce or amplify restriction; and loss-of-function, in which a CRISPR or shRNA or siRNA library is used in order to reveal restriction by disrupting it.
-
The laboratory of Charles M. Rice at Rockefeller University produced a number of gain-of-function screens to identify human ISGs inhibiting flaviviruses as well as viruses from other families. In 2011, their paper on the effects of hundreds of ISGs over-expressed in human fibroblasts on the infectivity of WNV and YFV, among other viruses, identified many inhibitory factors (Schoggins et al. 2011). Although most of the ISGs identified were activators of the whole IFN-I pathway (e.g. IRF1, a transcription factor for IFN-β) rather than genuine effectors, it also pointed to the anti-flaviviral effect of new factors such as IFN-α-inducible protein 6 (IFI6) and heparanase (HPSE) (Schoggins et al. 2011). A similar screen was performed in cells infected with DENV, also yielding IFI6 as well as additional hits (Schoggins et al. 2012). Earlier, the group of Ju-Tao Guo at Drexel University had used a similar approach to identify DENV and WNV restriction factors, but their screen was smaller and focused on antiviral effectors, hence excluding activators and regulators of the pathway (Jiang et al. 2010). This screen was highly successful, resulting in the identification of IFITM2, IFITM3, ISG20 and viperin as gene products inhibiting both viruses. IFITM2 and IFITM3 are well-known factors that restrict the fusion and/or disassembly of enveloped viruses belonging to several families, and they were shown to inhibit ZIKV and YFV in addition to WNV and DENV (Brass et al. 2009; Jiang et al. 2010; Schoggins et al. 2011; Gorman et al. 2016; Savidis et al. 2016). IFITM2 and IFITM3 were also flagged as potential flavivirus restriction factors in the Schoggins studies (Schoggins et al. 2011; Schoggins et al. 2012), but ISG20 and viperin were not, underlining the fact that apparent minor variations in the way a screen is built (cell line and virus used, infection readout, etc.) can have a significant impact on the results. Yamamoto and colleagues at National University of Singapore treated HeLa cells with IFN-I, then used mRNAs extracted from these cells to build a cDNA library which was transduced into Huh7 cells (Suzuki et al. 2016). Among the transduced cells, those surviving a DENV infection challenge were isolated and the HeLa mRNA over-expressed was sequenced. Most surviving clones expressed C19orf66, quickly renamed RyDEN (see below). Again, the fact that this screen yielded hits that were mostly not redundant with other screens testifies to the fact that highly experimental system-dependent results are to be expected in such screens. Recently, the group of Ella Sklan at Tel Aviv University performed a highly innovative CRISPR-based activation screen (Dukhovny et al. 2019). They used lentiviral vectors to transduce a single-guide RNA (sgRNA) library in human Huh7, along with a nuclease-inactivated Cas9 fused to a transcriptional activator (Konermann et al. 2015). The cells were then infected with ZIKV, and surviving cells were recovered after 10 days. The relative frequency of sgRNA in this population, compared to a control uninfected population, pointed to genes whose activation had protected the cells. Strikingly, IFI6 was isolated again, cementing its importance in controlling flaviviruses, along with ISG20 and additional hits, including some that are involved in virus sensing (MAVS, TRIM25) (Dukhovny et al. 2019).
-
The Fikrig laboratory silenced 21, 121 human genes by siRNA transfection, leading to the identification of mono-carboxylate transporter 4 (MCT4) as a putative restriction factor for WNV (Krishnan et al. 2008). The mechanism of inhibition, however, is elusive. The Diamond laboratory also performed a screen to isolate WNV-targeting restriction factors, which was limited to 245 human ISGs (Li et al. 2013). In this screen, the impact of ISG knockdown on WNV infection was measured in IFN-β-treated HeLa cells. Up to 16 putative WNV restriction factors were identified, most of them uncharacterized yet, with the notable exceptions of IFI6 and OAS1 (Li et al. 2013). Interestingly, many putative effectors inhibiting WNV in this screen also decreased infection by DENV (Li et al. 2013). In 2018, the Schoggins laboratory at University of Texas lentivirally transduced the flavivirus-permissive Huh7 human cell line with a CRISPR library, treated the cells with IFN-α, infected them with YFV and isolated the cells that were successfully infected despite the treatment (Richardson et al. 2018). Among the ~15 hits obtained, only one was an antiviral effector, IFI6, whereas most of the other hits were pathway activators/regulators. The same group performed a detailed analysis of the mechanism of flaviviral restriction by IFI6 (see below).
Thus, the loss-of-function and gain-of-function approaches each yielded interesting and mostly nonredundant restriction factor candidates, some of them poorly studied to this day (e.g. ISG20). The genetic screens performed so far used wild-type human viruses in human cells. It is reasonable to assume that additional restriction factors might be discovered in the future, that were missed in the studies cited above due to the fact that flaviviruses are adapted to their human host and thus probably escape some intrinsic responses. In this regard, the history of retrovirus restriction factor discovery is informative. Indeed, many of them were identified through the use of infection-permissive vs non-permissive cells (e.g. TRIM5α, APOBEC3G) (Sheehy et al. 2002; Stremlau et al. 2004), or by using mutant viruses that had lost the capacity to escape restriction (e.g. Tetherin, SERINC5) (Neil et al. 2008; Usami et al. 2015). Because most restriction factors establish direct contacts with their viral targets, another possible approach consists in purifying cellular interactors for a given viral protein, and then to analyze whether some of these interactors might be restriction factors. For instance, Taylor and colleagues used the yeast-two-hybrid system to look for cellular proteins interacting with the NS5 protein of LGTV; this led to the identification of murine TRIM79α as a LGTV and TBEV restriction factor, as detailed below (Taylor et al. 2011).
Gain-of-Function Screens
Loss-of-Function Screens
-
As mentioned above, IFI6 was isolated in multiple gain-of-function and loss-of-function genetic screens for flaviviral restriction factors, underlining its relevance. IFI6 inhibits all flaviviruses tested so far (YFV, WNV, DENV, ZIKV), and contributes to the IFN-I antiviral effect against these viruses (Richardson et al. 2018; Dukhovny et al. 2019). IFI6 does not affect entry nor early post-entry translation, but it does delay RNA replication and decreases late translation of viral genes (Richardson et al. 2018). Because IFI6 is a signal peptide-containing, hydrophobic protein localizing at the secretory system and more specifically in the membranes of the ER (Richardson et al. 2018; Dukhovny et al. 2019), investigators analyzed its effect on the formation of flavivirus replication complexes. They found that IFI6 overexpression prevented YFV-induced ER invaginations that are characteristic of replication organelles (Arakawa and Morita 2019). These data suggest that IFI6 acts indirectly by preventing the establishment of favorable replication conditions for flaviviruses (Fig. 1), and may not make any direct contact with flaviviral molecules. This lack of direct targeting requirement, which remains to be confirmed, would also explain why IFI6 seems to inhibit all flaviviruses. The Schoggins group also reported that IFI6 antiviral activity required another ER protein, the heat shock protein family member BiP (also called GRP78, and encoded by HSPA5), which they also isolated in a genetic screen (Richardson et al. 2018). This finding, however, seems to contradict previous reports that BiP has a positive role in the flavivirus life cycle (Lewy et al. 2017).
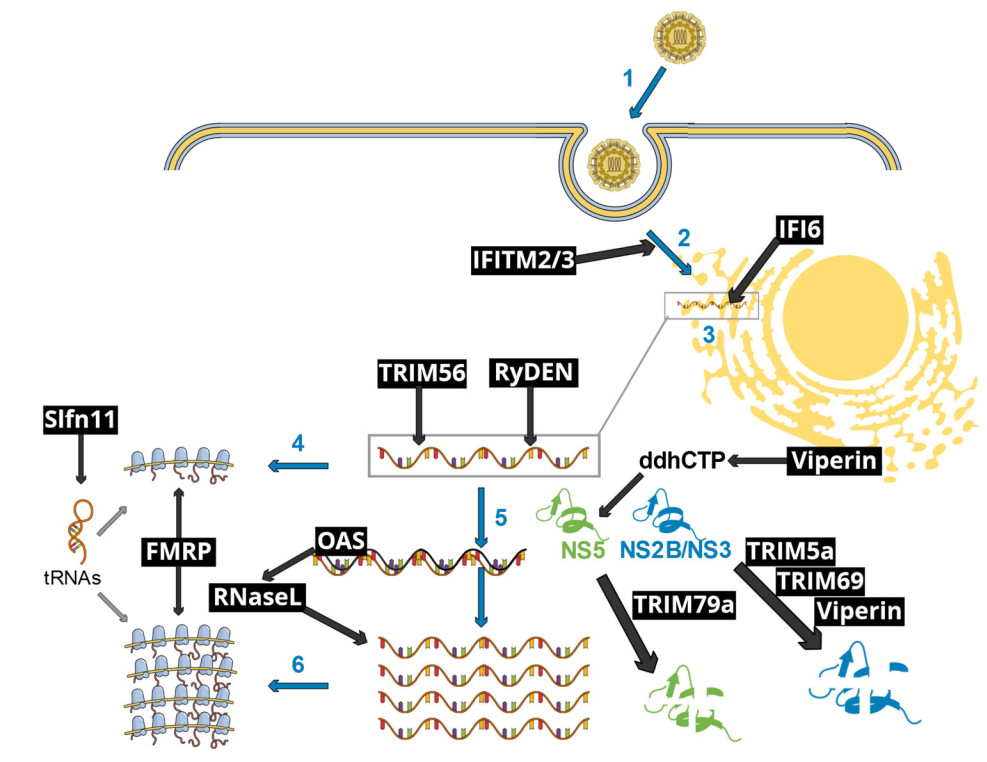
Figure 1. Schematic representation of early flavivirus infection stages affected by selected IFN-I-inducible restriction factors. Blue arrows and numbers represent essential viral infection steps, as follows: (1) flavivirus particle attachment to its specific receptor, followed by endocytosis-mediated entry; (2) fusion accompanied with uncoating of the viral core; (3) establishment of replication organelles at the endoplasmic reticulum, where both genome replication and gene expression occur; (4) early translation of the incoming viral genome, generating the viral enzymes necessary to viral replication, such as the protease NS2B/NS3 and the RNA polymerase NS5; (5) viral genome replication through double-stranded intermediates; (6) late viral translation, which yields the structural proteins for the subsequent assembly of novel viral particles (not shown). Black text boxes and arrows show restriction factors and pathways. IFITM2/3 interfere with fusion and uncoating. IFI6 prevents the establishment of replication organelles, thus disrupting viral RNA replication. TRIM56 and RyDEN bind to the viral RNA, affecting RNA replication. OAS proteins activate RNaseL upon binding to double-stranded RNA replication intermediates, resulting in the digestion of both viral and cellular RNAs. Viperin promotes the formation of a nucleotide analog, ddhCTP, resulting in the inhibition of the NS5-mediated genome replication. NS5 is targeted to a lysosomal degradation pathway by TRIM79α. The viral NS2B/NS3 protease is targeted to a proteasomal degradation pathway by TRIM5α, TRIM69 as well as Viperin. FMRP inhibits viral gene-translating ribosomes. Finally, Slfn11 reduces the levels of key tRNAs, hence inhibiting viral gene translation. This schematic view does not take into account virus-specific, host species-specific and cellular context-specific limitations to the restriction pathways shown.
-
Viperin (virus-inhibiting protein, endoplasmic reticulum-associated, interferon-induced), also called radical S-adenosyl methionine domain containing 2 (RSAD2) and encoded in humans by RSAD2, localizes to the cytosolic side of the endoplasmic reticulum, which is also the site of flavivirus replication. Viperin has orthologs in other mammal species, such as the mouse, and homologs in nonmammal vertebrates such as fish (Chin and Cresswell 2001), arguing for an evolutionarily conserved role. The central domain binds an iron-sulfur cluster, and has radical S-adenosyl-L-methionine (SAM) catalytic activity (Duschene and Broderick 2010). Upon addition of SAM to viperin, the former is cleaved to produce 5'-deox-yadenosine, a reaction characteristic of the radical SAM family of enzymes. Viperin has been shown to inhibit multiple RNA viruses, belonging to different families (Fitzgerald 2011). However, it was first discovered as a restriction factor for a DNA virus, the human cytomegalovirus (HCMV) (Chin and Cresswell 2001). Viperin also reduces permissiveness to infection by a lentivirus, HIV-1 (Rivieccio et al. 2006), as well as HCV, a Hepacivirus from the Flaviviridae family (Helbig et al. 2005; Jiang et al. 2010) and influenza A virus (IAV), an orthomyxoviridae (Wang et al. 2007). The first demonstration of an antiviral effect against flaviviruses came in 2010 (Jiang et al. 2010). In a murine model of WNV, expression of viperin was key to controlling viral replication in the central nervous system, significantly reducing lethality (Szretter et al. 2011). Human viperin efficiently inhibits DENV in a variety of human cell lines and primary macrophages as investigated by both depletion and overexpression (Helbig et al. 2013). It inhibits DENV at an early post-entry stage by interfering with the generation of minus-strand viral genomic RNA which is copied from the plus-strand by the viral NS5 polymerase (Helbig et al. 2013). Similarly, viperin strongly inhibited the replication of TBEV and ZIKV in cell cultures, and similar to DENV, the block was post-entry and affected RNA synthesis by the viral polymerase (Upadhyay et al. 2014; Vanwalscappel et al. 2018).
Protein-RNA co-immunoprecipitation and immunofluorescence microscopy approaches suggested that viperin was present at DENV RNA replication complexes (Helbig et al. 2013). Similarly, viperin was shown to co-localize with HCV replication complexes, and the C-terminal region of viperin had a role in its localization (Helbig et al. 2005). HCMV infection of human cells recruited viperin to viral replication and production sites at the Golgi apparatus and vacuoles ("droplets")containing new viral particles (Chin and Cresswell 2001). The N-terminal region of viperin contributes to its anchoring into lipidic intracellular membranes (Hinson and Cresswell 2009a, Hinson and Cresswell 2009b). In summary, viperin inhibits multiple families of RNA and DNA viruses, and its recruitment to viral replication sites is likely to be important for this inhibitory process.
The molecular mechanism by which viperin inhibits multiple family of viruses was poorly understood for more than 15 years. It was proposed to decrease virus release by perturbing lipid rafts (Wang et al. 2007), but this mechanism was not consistent with the early effects observed on flaviviral replication. The central iron-sulfur-binding catalytic domain was found to be necessary to TBEV restriction, pointing to the importance of the viperin catalytic activity (Upadhyay et al. 2014). A quantum leap forward was achieved in 2018, when the Steven Almo group at Albert Einstein College of Medicine demonstrated that viperin catalyzes the generation of a nucleotide analog. Specifically, it converts cytidine triphosphate (CTP) to 3'-deoxy-3', 4'-didehydro-CTP (ddhCTP) (Gizzi et al. 2018). IFN-I-mediated transcriptional upregulation of viperin is accompanied by the upregulation of cytidylate monophosphate kinase 2 (CMPK2), which converts CDP to CTP, hence increasing the amount of substrate for viperin. ddhCTP inhibits the RNA-dependent RNA polymerase activity of DENV, WNV, ZIKV and hepatitis C virus by competing with CTP for incorporation into de novo-synthesized RNA in cell-free assays (Gizzi et al. 2018). Thus, ddhCTP acts as a chain terminator, similar to the mechanism of action of many pharmaceutical nucleoside analogs used to treat viral infections (von Kleist et al. 2012). Importantly, the same group treated ZIKV-infected cells with ddhC, the nonphosphorylated form of ddhCTP (only non-phosphorylated nucleotides efficiently cross the plasma membrane, and are subsequently phosphorylated in the cell). Such treatment inhibited the replication of ZIKV, providing a proof-of-concept for pharmacological applications (Gizzi et al. 2018). Also in 2018, viperin was found to disrupt replication of TBEV and another flavivirus, Langat virus (LGTV) by promoting the release of noninfectious capsid particles (Vonderstein et al. 2018). Whether this phenotype is a byproduct of the RNA replication inhibition or whether it constitutes a distinct function of viperin remains to be determined. Other mechanisms of action have been reported for viperin, such as the binding to and proteasomal degradation of TBEV and ZIKV NS3 protease (Panayiotou et al. 2018). Whether viperin inhibits flaviviruses through several simultaneous mechanisms of action remains to be determined.
-
The human genome contains more than 70 genes known to encode TRIM proteins. TRIM proteins are characterized by the presence of a RBCC tripartite motif composed of a RING (really interesting new gene) domain with E3 ubiquitin ligase activity, one or two B-box domains and a Coiled-coil domain (Reymond et al. 2001; Hatakeyama 2017). The C-terminal region is more variable, providing the basis for the classification of TRIM proteins in 11 subfamilies (Short and Cox 2006; Ozato et al. 2008). TRIM proteins are involved in a number of cellular functions, but their role in cellular antiviral immunity has emerged as crucial in the last 15 years (Ozato et al. 2008; van Gent et al. 2018). TRIM5α, encoded by TRIM5, is an IFN-I-activated, SPRY (PRYSPRY, B30.2)-containing group IV TRIM protein (Ozato et al. 2008; Carthagena et al. 2009), whose antiviral activity targeted at retroviruses was discovered in 2004 (Stremlau et al. 2004; Ganser-Pornillos and Pornillos 2019). TRIM5α interacts as a multimer with the retroviral capsid core early after virus entry into the cell, trapping them in cytoplasmic bodies and inducing disassembly of the core (Campbell et al. 2008; Roa et al. 2012). The E3 ubiquitin ligase of the RING domain is necessary for the disruption of the retroviral core, but is not required for retrovirus sequestration in TRIM5α cytoplasmic bodies (Campbell et al. 2008). Direct interactions between hyper-variable loops in the TRIM5α C-terminal SPRY domain and the retroviral capsid N-terminal domain constitute the molecular basis for the restriction (Sawyer et al. 2005). Recently, human TRIM5α was also found to restrict the tick-borne flaviviruses TBEV, LGTV and Kyasanur Forest disease virus (KFDV) in a study from the Best laboratory (Chiramel et al. 2019). Restriction was shown both using TRIM5α overexpression and conversely by reducing expression using RNAi and CRISPR. Importantly, TRIM5α contributed to the IFN-β-induced antiviral response against those same viruses. The inhibition was characterized by a decrease in viral RNA replication. TRIM5α interacts with the viral protease heterodimer NS2B/NS3, leading to the poly-ubiquitination and proteasome-mediated degradation of the protease (Chiramel et al. 2019). TRIM5α partially colocalizes with NS2B/NS3 in infected cells, and the interaction is also seen upon virus-free expression of the protease (Chiramel et al. 2019). The TRIM5α RING ubiquitin ligase domain is necessary for the proteasome-mediated degradation of NS2B/NS3, whereas the C-terminal SPRY domain is responsible for TRIM5α targeting of the viral target (Chiramel et al. 2019). Strikingly, these TRIM5α domains have similar functions in the restriction of retroviruses (Ganser-Pornillos and Pornillos 2019). Future studies will probably map with greater precision the motifs of TRIM5α and NS2B/NS3 required for interaction and restriction, which might explain why some flaviviruses are not sensitive to this restriction factor. Nonetheless, this work by Chiramel and colleagues provides ground-breaking evidence that TRIM5α has evolved to target two unrelated families of RNA viruses.
-
Similar to TRIM5α, TRIM69 is a SPRY domain-containing, IFN-I-activated group IV TRIM protein (Ozato et al. 2008; Carthagena et al. 2009). TRIM69 was known mostly for its role in zebrafish brain development (Han et al. 2016) until the group of Jianfeng Dai at Soochow University demonstrated its function as a DENV restriction factor (Wang et al. 2018). TRIM69 was investigated due to it being one of the ~100 mRNAs induced following infection of 293T cells with DENV. Using both overexpression and knockdown, they show that TRIM69 decreases DENV infectivity and contributes to the inhibitory effect of IFN-I (Wang et al. 2018). They also performed elegant in vivo TRIM69 knockdown experiments using lentiviral vectors, demonstrating that TRIM69-depleted mice were significantly more susceptible to infection by DENV (Wang et al. 2018). Mechanistically, TRIM69 directly interacts with NS3 and promotes its polyubiquitination and degradation. Accordingly, the RING domain-associated E3 ubiquitin ligase activity was required for restriction. Whether TRIM69 targets other flaviviruses is not known yet.
-
Mus musculus TRIM79α (NP_954597), encoded by Trim30d, does not have an ortholog in the human genome, but its closest human homologs are SPRY domain-containing Group IV TRIM proteins, most notably TRIM5α (45.7% amino acid identity), TRIM22 (43.1%), and TRIM34 (42.1%). TRIM79α, whose expression is induced by IFN-I treatment or exposure to viruses, was found in 2011 to inhibit the tick-borne flaviviruses TBEV and LGTV (Taylor et al. 2011). Although this discovery is not recent, it is interesting to compare the mechanism of restriction to that of human TRIM5α. Whereas TRIM5α targets the protease, TRIM79α targets the NS5 polymerase. Whereas TRIM5α-induced degradation is mediated by the proteasome, TRIM79α uses lysosomes, albeit the study was limited in terms of cell types (most experiments were performed in 293 cells). Consistent with the lysosomal degradation, TRIM79α anti-flaviviral activity did not require a functioning RING domain (Taylor et al. 2011), but was inhibited by treatment with a drug targeting lyso-somes. It would be interesting to determine whether there is a functional human homolog to TRIM79α, i.e. a human TRIM protein directing the lysosomal degradation of NS5 (Box 1).
Flavivirus restriction vs. sensing
● Do some flavivirus restriction factors also function as sensors?
IFI6
● Does IFI6 engage in direct contacts with flaviviral targets?
Viperin
● Is the enzymatic activity of viperin constitutive or triggered by interactions with viral motifs?
● Do mammalian cells encode other enzymes involved in the synthesis of natural nucleotide analogs with antiviral activity?
● Could the antiviral effect of the viperin enzymatic activity serve as a guide toward the development of novel nucleoside analog pharmaceuticals?
Cytoplasmic TRIMs
● What are the precise viral and TRIM molecular determinants of flavivirus restriction specificity?
● Is there a functional human homolog of TRIM79α able to target the NS5 protein of flaviviruses toward a degradation pathway?
● Is the restriction of incoming flaviviruses by cytoplasmic TRIMs a coordinated response involving TRIM-TRIM interactions?
RNA-targeting factors
● Are there functional interactions between the various flaviviral RNA-binding restriction factors?
● What is the basis for selective flavivirus species RNA recognition by human OAS proteins?
● Does human ABCF3 contribute to OAS-mediated restriction of flaviviruses?
TRIM19 (PML)
● What is the mechanism by which TRIM19 decreases susceptibility to DENV?
Escape from restriction
● Do flaviviruses escape some restriction factors through direct targeting by viral proteins?
● Do flaviviruses other than DENV disrupt TRIM19 nuclear bodies?Table Box 1. Unanswered questions in the restriction of flaviviruses.
-
The Group V TRIM protein TRIM56 is a cytoplasmic protein whose expression is moderately stimulated by IFN-I, and was first described as a restriction factor for the bovine viral diarrhea virus (BVDV), a member of the pestivirus genus of the Flaviviridae (Wang et al. 2011). The C-terminal domain of TRIM56 contains NHL-like domains that have unclear molecular function but were required for the restriction of BVDV (Wang et al. 2011). More recently, the Kui Li laboratory performed human TRIM56 overexpression and knockdown experiments to show that it restricted DENV as well as YFV (Liu et al. 2014). Both a functional RING domain and the presence of the C-terminal NHL-like repeats were required for TRIM56 to restrict these viruses (Liu et al. 2014). The molecular target was not identified, but restriction was associated with a decrease in RNA replication, suggesting that, like TRIM5α and TRIM79α, TRIM56 targets the viral replication complexes. Recently, the same group followed up on these discoveries by showing that TRIM56 also restricted ZIKV (Yang et al. 2019). Similar to DENV and YFV, ZIKV restriction involved both the RING domain and NHL-like repeats, and restriction was associated with a decrease in viral RNA replication (Yang et al. 2019). Pulldown experiments showed that TRIM56 interacted with the ZIKV viral RNA, and that the NHL-like repeats but not the RING domain were required for this interaction (Yang et al. 2019). Additional cell-free, virus-free experiments suggested that the TRIM56-RNA interaction was direct rather than the result of an interaction between TRIM56 and a viral protein (Yang et al. 2019).
Altogether, the TRIM5α, TRIM69, TRIM79α and TRIM56 studies show that cytoplasmic TRIMs restrict flaviviruses through a common scheme that involves a direct interaction between the TRIM C-terminal domain and a viral target that may be a protein or RNA. The TRIM RBCC domains implement the restriction itself, through poorly known mechanisms involving the RING E3 ubiquitin ligase activity or not. Almost 20 years ago, Reymond and colleagues showed that TRIM proteins were prone to both homo and heterodimerization (Reymond et al. 2001), a phenomenon that has received little attention since. It is conceivable that cytoplasmic antiviral TRIM proteins might interact with one another and form complexes that restrict flaviviruses by targeting multiple viral components simultaneously (Box 1).
-
RyDEN (repressor of yield of DENV), also called IRAV (interferon-regulated antiviral gene) and encoded by SHFL, was identified by the Yamamoto group at National University of Singapore, as a DENV restriction factor in a gain-of-function genetic screen. Specifically, it was isolated as an IFN-α-stimulated HeLa mRNA protecting Huh7 cells from DENV infection (Suzuki et al. 2016). Another team reached similar conclusions the following year (Balinsky et al. 2017). A detailed analysis by the two groups showed that virus entry is not affected, but RNA replication is decreased. RyDEN co-localizes with DENV replication complexes, and binds to the DENV RNA as shown in biochemical experiments performed in cell lysates or using purified molecules (Suzuki et al. 2016; Balinsky et al. 2017). RyDEN also inhibits unrelated RNA and even DNA viruses (Suzuki et al. 2016; Rodriguez et al. 2019), suggesting that it has broad antiviral activity through the targeting of viral genomes or via additional mechanisms.
-
2', 5'-oligoadenylate synthetase (OAS), which is induced by IFN-I, cooperates with RNase L to inhibit a number of RNA viruses in one of the most well-characterized antiviral cell-autonomous antiviral pathways (reviewed in (Silverman 2007)). In this pathway, OAS enzymes recognize dsRNA structures, characteristic of RNA virus replication intermediates, or stem-loops present in the flaviviral genome (Deo et al. 2014). The OAS-RNA interaction stimulates OAS to catalyze the oligomerization of ATP into 2', 5'-linked oligoadenylate, which in turn binds to and activates latent RNase L endonucleases (Fig. 1). RNase L cuts both viral and cellular RNA, decreasing virus production and potentially affecting cell viability (Gusho et al. 2016; Banerjee et al. 2019). A nonsense mutation in one of the isoforms of the murine OAS (OAS1b) was shown to confer mice susceptibility to WNV infection (Mashimo et al. 2002; Perelygin et al. 2002). Accordingly, RNase L was demonstrated to contribute to the resistance of mice and murine cells to infection by WNV (Samuel et al. 2006). It has also been proposed that some OAS isoforms could inhibit RNA viruses independently of RNase L, perhaps through a putative nuclease activity associated with their C-terminal domain (Rogozin et al. 2003). Four OAS genes are found in humans, encoding a total of 10 isoforms (Mashimo et al. 2003; Lin et al. 2009). In vitro, the isoforms OAS1 p42, OAS1 p46 and OAS3 restricted DENV upon overexpression (Lin et al. 2009). Restriction was strictly dependent upon the presence of RNase L, and degradation of cellular RNA was seen (Lin et al. 2009). Conversely, knocking out RNase L increased DENV and ZIKV genome replication in human cells (Whelan et al. 2019). Thus, DENV and ZIKV are targeted by the OAS/ RNase L pathway in human cells. Interestingly, ZIKV appears to be partially resistant to RNase L activation, but resistance is overcome by pre-treating the cells with synthetic dsRNAs, suggesting that ZIKV is not efficiently detected by the human OAS enzymes (Whelan et al. 2019). Deciphering the molecular mechanisms that govern the susceptibility of flaviviruses to detection by human OAS enzymes is key to a fuller understanding of this restriction pathway (Box 1). Additional partners in this pathway have been suggested, such as the little-studied ABCF3 (ATP binding cassette subfamily F member 3), part of a large family of membrane-bound ATP-binding transporter proteins (ABC transporters). ABCF3 was found to physically interact with the murine OAS1b and to act as a WNV restriction factor in murine cells (Courtney et al. 2012). Recently, ABCF3 was shown to have ATPase activity, though how this activity is connected to the antiviral effect is at present unclear (Peterson et al. 2019). Whether the human ABCF3 ortholog could cooperate with one of the human OAS isoforms to target some flaviviruses is also not known.
-
FMRP is an IFN-I-stimulated (source: interferome.org) cytoplasmic protein expressed at high levels in brain cells and important for neurodevelopment (Davis and Broadie 2017). FMRP was recently isolated in a screen for cellular proteins binding to ZIKV subgenomic flaviviral RNAs (sfRNAs), which are RNAs produced by the viral replication machinery that do not contribute to synthesizing viral proteins and are noninfectious, but nonetheless accomplish important functions for flaviviruses (Mazeaud et al. 2018). FMRP bound both genomic RNA and sfRNAs, but binding to the latter was more efficient (Soto-Acosta et al. 2018). FMRP knockdown increased the permissiveness of HeLa cells to infection by several ZIKV strains but not DENV (Soto-Acosta et al. 2018). The magnitude of FMRP-mediated restriction was greater for A10 ZIKV, a vaccine strain that produces less sfRNAs (Shan et al. 2017). Restriction did not affect viral RNA replication but was accompanied with a decrease in viral protein translation (Soto-Acosta et al. 2018). Therefore, it is proposed that FMRP is a restriction factor targeting ZIKV-translating ribosomes that is counteracted by ZIKV sfRNAs perhaps through a "sponge" mechanism (Soto-Acosta et al. 2018). This restriction mechanism is consistent with the known affinity of FMRP for ribosomes (Chen et al. 2014). It remains to be seen whether this restriction activity is present in different cell types, and whether it is significant in vivo. However, together with OAS, Ryden and TRIM56, the FMRP findings underline genomic RNA as a major flaviviral molecule recognized and targeted by cytoplasmic restriction factors (Fig. 1).
-
PML is an IFN-I-stimulated nuclear group V TRIM protein (Grotzinger et al. 1996; Ozato et al. 2008), long known to be involved in the cellular defenses against viruses and in particular to modulate the infectivity and latency potential of herpesviridae (Everett et al. 2008; Gunther et al. 2014). It forms spherical nuclear bodies that host a number of additional proteins. PML is heavily conjugated to SUMO (small-ubiquitin like modifier) proteins, and many of the additional proteins localizing at PML nuclear bodies are also SUMOylated (Lallemand-Breitenbach and de The 2018). It has emerged that PML and PML nuclear bodies inhibit the infectivity of multiple DNA and RNA viruses indirectly through the transcriptional regulation of the IFN-I response (Kim and Ahn 2015; Scherer and Stamminger 2016). Knockdown of PML in A549 cells leads to an increase in susceptibility to infection by DENV (Giovannoni et al. 2015). Conversely, overexpression of one of the PML isoforms (isoform IV) reduces DENV infectivity (Giovannoni et al. 2015). The mechanism of restriction is unknown, but because PML is almost exclusively nuclear, it is likely to be indirect/regulatory, consistent with the role of PML in modulating the expression of ISGs (Xu et al. 2009; Kim and Ahn 2015). Of note, PML also negatively modulates the cytoplasmic replication stages of retroviruses (Masroori et al. 2016).
-
The Schlafen family of proteins is involved in the regulation of important cellular functions such as the regulation of DNA repair and the inhibition of viral replication (Mavrommatis et al. 2013). Five Schlafen genes have been identified in humans, of which at least four (Slfn5, 11, 12 and 13) are upregulated by IFN-I (source: Interferome.org). Slfn11 and Slfn13 are endonucleases that target specific tRNAs, resulting in the modulation of specific sets of cellular or viral genes (Yang et al. 2018; Malone et al. 2019). It was shown in 2012 that Slfn11 is a restriction factor for HIV-1, which was explained by the specificities of HIV-1 codon usage (Li et al. 2012). Very recently, the group of Manuel Llano at University of Texas showed that Slfn11 also restricted WNV, DENV and ZIKV in human glioblastoma A172 cells (Valdez et al. 2019). Restriction was clearly cell context-specific, as no inhibition of WNV was seen upon overexpression of Slfn11 in three other cell lines (HeLa, HEK293T and BHK-21). As expected, Slfn11 expression affected the relative concentration of sensitive tRNAs (Valdez et al. 2019). The presence of Slfn11 decreased the amounts of infectious particles in the supernatants, but the link between this and the degradation of specific tRNAs is not clear yet. Future work will probably shed light on whether viral protein translation is directly affected by Slfn11-mediated regulation of tRNA pools, and will explain the cell context specificity.
IFI6
Viperin
TRIM5α
TRIM69
TRIM79α
TRIM56
RyDEN (IRAV, C19orf66)
OAS, RNase L and ABCF3
Fragile X Mental Retardation Protein
TRIM19 (PML)
Schlafen 11
-
That viruses evolve mechanisms to counteract the cell-autonomous immune response is not only expected, but also constitutes proof of the in vivo relevance of these innate pathways. Similar to other classes of viruses, evidence has been uncovered of flavivirus interference with PRR-mediated sensing of the virus, with IFN-I production and with the antiviral pathways downstream of IFN-I binding to its receptor (Reviewed in (Diamond 2009; Miorin et al. 2017)). Sensing of DENV by RIG-I is decreased through the sequestration of TRIM25 by sfRNAs (Manokaran et al. 2015). The NS2B/NS3 protease of DENV and ZIKV directly cleaves the signal transducer STING, damping IFN-I production (Aguirre et al. 2012; Ding et al. 2018). Ectopic overexpression of DENV proteins NS2A, NS4A or NS4B blunts the antiviral state stemming from treatment with IFN-I (Munoz-Jordan et al. 2003). NS4B acts by inhibiting the phosphorylation of STAT1 that occurs downstream of IFN-I binding to its cellular receptor (Munoz-Jordan et al. 2003). The NS5 protein of JEV, but not its NS4B protein, similarly prevents the phosphorylation and nuclear import of STAT1 following IFN-α treatment (Lin et al. 2006). Similar to viruses belonging to other families, flaviviruses may have evolved mechanisms to interfere with the antiviral functions of PML and PML bodies. Indeed, infection of A549 cells with DENV resulted in a decrease in the number of PML bodies (Giovannoni et al. 2015), but the mechanism and viral protein(s) involved in this interference effect are unknown. It also remains to be seen whether this inhibition of PML bodies can be generalized to other flaviviruses.
Thus, there is ample evidence of flaviviral proteins and RNA interfering with IFN-I-related antiviral pathways. Evidence of flavivirus escape from individual restriction factors, on the other hand, is scarce. This might be because flaviviruses encode a small number of proteins (10), which limits their potential for evolving restriction factor-specific responses. In other words, it is probably more cost-efficient for flaviviruses to interfere with the IFN-I antiviral pathway globally than it would be to counteract individual antiviral effectors. Nonetheless, cases of flavivirus escape from specific restriction factors have been documented. Perhaps the most well-understood mechanism is the role of flaviviral NS5 2'-O-methyltransferase activity that modifies the capped 5' extremity of the viral genome, hence avoiding restriction by the IFIT family of proteins (Daffis et al. 2010). Binding of IFIT (interferon-induced with tetratricopeptide repeats) proteins to uncapped RNA extremities prevents translation of the viral proteins (Fensterl and Sen 2015), but the methylated extremity is not recognized. Accordingly, WNV and JEV bearing mutations in the 2'-O-methyltransferase domain of NS5 are hypersensitive to IFN-I (Szretter et al. 2012; Kimura et al. 2013). Viperin is also suspected of being counteracted by a viral factor. Comparison of the effect of viperin on two viruses, Sindbis virus (SINV, from the Togaviridae family) and the flavivirus JEV revealed the existence of an escape mechanism by the latter, based on the proteasome-mediated degradation of viperin and associated with unidentified viral factors (Chan et al. 2008). Accordingly, viperin inhibited both viruses in the presence of the proteasome inhibitor MG132 (Chan et al. 2008). Finally, and as explained above, sfRNAs allow ZIKV to resist restriction by FMRP. It is not know yet, however, whether any flaviviral protein makes direct contact with the host restriction factor in order to inactivate it (Box 1).
-
In one of the most interesting developments in the field of retrovirus restriction factors, TRIM5α and Tetherin (BST2) were found to activate innate pathways following their binding to viral targets (Pertel et al. 2011; Hotter et al. 2013; Merindol et al. 2018). Hence, these effectors also act as PRRs. Could it also be the case for some flavivirus restriction factors? In a murine model for WNV infection, the presence of viperin moderately increased the early IFN-I response, as seen by IFN-I concentration in peripheral blood (Szretter et al. 2011). In viperin-knockout murine cells, production of IFN-α and IFN-β was reduced compared to wild-type cells, following stimulation with TLR7 ligands (heat-treated Newcastle disease virus or R848) or TLR9 ligands (CpG oligodeoxynucleotides) (Saitoh et al. 2011). It is not known yet whether viperin is involved in flavivirus sensing concomitantly to its restriction activity, and its role in TLR7-and TLR9-mediated sensing has not yet been investigated in human cells. However, viperin recruits signal mediators IRAK1 and TRAF6 to cytoplasmic lipid droplets, suggesting that it might contribute to flavivirus sensing by attracting the sensing machinery to the viral replication sites (Saitoh et al. 2011).
-
TLR agonists are used to boost the immune response to DNA viruses. Resiquimod and Imiquimod, two related compounds that act as TLR7/8 agonists, are used to treat genital herpes caused by Herpes simplex virus type 2 (HSV-2) and genital warts caused by human papillomavirus (HPV), respectively (Mark et al. 2007; Gotovtseva et al. 2008). Accordingly, the TLR7/8 agonist R848 stimulated viperin expression in primary macrophages (Vanwalscappel et al. 2018). Boosting the intrinsic response against flaviviruses constitutes a promising direction for future therapeutic agents.
-
Knowledge of the control of flaviviral infection by IFN-I-dependent effectors is rapidly expanding, but much remains to be discovered. Similar to retroviruses, flaviviruses are targeted intracellularly at multiple steps of their life cycle. NS3, NS5 and the viral RNA appear to be privileged targets for restriction factors, but this observation may be biased by the fact that other viral proteins have received little attention so far. Some restriction factors (e.g. IFI6) may target all flaviviruses tested, but the vast majority show some level of virus specificity. Understanding selective viral sensitivity to restriction will require a deeper knowledge of the molecular determinants governing the detection of the viral protein or RNA targets by the restriction factors. Genetic screen approaches to discover flavivirus restriction factors have been highly successful. However, readout for these screens was based upon the detection of a fluorescent marker expressed from the viral genome, or upon survival of the cells. Because of this, the restriction factors isolated so far tend to target early stages of the infection, rather than disrupt the late stages of the virus life cycle such as virus assembly and release. In the future, species-specific and context-specific differences in susceptibility to the propagation of flaviviruses may perhaps provide fertile grounds for the development of genetic screens aimed at identifying novel restriction factors, including late stage-targeting factors. Finally, at least two of the antiviral effectors discussed here (TRIM19 and Slfn11) have no known flaviviral targets and probably act through indirect mechanisms, which may also be the case for IFI6. This raises the question of whether they should be considered as belonging to a subgroup of restriction factors, defined as factors depriving the virus of an optimal environment, rather than directly attacking viral targets. Among the major open questions in flavivirus restriction is whether some flaviviral proteins can directly inactivate restriction factors, as has been repeatedly observed for retroviruses. Flaviviruses are increasingly recognized as a major international threat, owing to their frequent involvement in outbreaks and the scarcity of drugs and vaccines available to combat them. A better understanding of restriction factors may pave the way toward novel therapies against flaviviruses, for instance in the form of small molecules designed to activate specific restriction factors or modulate their activity. Undoubtedly, the next decade promises to deliver exciting advances in our knowledge of the flavivirus restrictome.
-
I apologize to colleagues whose excellent works I did not include in this review.
-
The author declares that he has no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by the author.

 DownLoad:
DownLoad: