














HTML
-
Ticks are one of the most important vectors being notorious for spreading etiological agent from wild hosts to domestic animals and humans. Since the first identification of Louping ill virus in ticks, which can cause severe encephalitis in sheep and other livestock (Stockman 1918), a large group of tick-borne viruses (TBVs) have been recognized, presenting great genetic diversity and various properties. Recently, the whole-genome sequencing of six ixodid tick species from China has been completed revealing rich variety of tick-associated pathogens (Jia et al. 2020). So far, TBVs are involved in at least 12 genera of nine families and two orders, and also include members that haven't been assigned (Shi et al. 2018).
TBVs that can replicate and maintain in ticks and infest vertebrate hosts pose severe threat to animal and human health (Horak et al. 2002). In China, it has long been plagued by TBVs including tick-borne encephalitis virus (TBEV) and Crimean-Congo hemorrhagic fever virus (CCHFV) in the northern provinces where fatal cases in several epidemics and sporadic outbreak were noted with a very high mortality rate decades ago (Moming et al. 2018; Zhang et al. 2012). In recent years, a novel TBV called severe fever with thrombocytopenia syndrome virus (SFTSV) has been identified in China, which is the causative agent of severe fever with thrombocytopenia syndrome (SFTS) disease to humans with a high initial mortality due to the lack of specific drugs and vaccines (Zhang et al. 2017). From 2010 to 2016, over 10, 000 SFTS cases have been reported in 23 provinces of China (Zhan et al. 2017). In 2018, China suffered an unprecedented livestock disaster implicating over 5000 pigs caused by African swine fever virus (ASFV) which transmitted among pigs, wild boar via soft ticks (Wang et al. 2018). So it is of great significance to investigate the tick-borne viromes in natural habitat for better control and prevention of large epidemic caused by TBVs.
The next-generation sequencing (NGS) has become a powerful technique used for investigating viromes of many species. Numerous novel viruses and virus-related sequences have been found from various arthropods using NGS, which largely boosted the current viral community (Tokarz et al. 2014; Li et al. 2015; Brinkmann et al. 2018). Novel viruses have also been identified or isolated from different tick species in recent years, indicating that the current knowledge of TBVs may be a tip of the iceberg.
Yunnan Province locates in the southwest of China, bordering on Myanmar, Laos and Vietnam. It is a quite suitable habitat for ticks in most areas of Yunnan Province, because of the abundant rainfall, lush vegetation, diverse wildlife and livestock (Xia et al. 2015). A variety of tick species have been identified in Yunnan Province, where Haemaphysalis and Rhipicephalus spp. ticks are commonly found (Chen et al. 2008). The Rhipicephalus (Boophilus) microplus tick is widely distributed in the world, and is an important vector for many viral pathogens, including Thogoto virus (Orthomyxoviridae, genus Thogoto), Wad Medani virus (Reoviridae, genus Orbivirus), CCHFV (Nairoviridae, genus Orthonairovirus), and Kandam virus (Flaviviridae, genus Flavivirus), which can cause diseases in livestock or humans (Labuda and Nuttall 2004). A number of researches have reported the viral diversity of R. microplus from different locations, such as Brazil and Colombia. And both common virus as well as region specific virus were identified (Gómez et al. 2020; Souza et al. 2018; Zhao et al. 2020). Previous study on R. microplus collected from different host shows that comparing to natural host of tick, the geographic location has more influence on virus profile (Zhao et al. 2020). R. microplus is also the dominant tick species in Yunnan Province, however, its virome profiling has not been fully characterized. In this study, R. microplus ticks were collected from domestic cattle surface from 2015 to 2017 in Yunnan Province. We investigated the virome of R. microplus using NGS and the prevalence of important identified viruses among tick groups by RT-PCR. Serological survey was performed to reveal the potential transmission and infection of these identified viruses to human and cattle. The data will contribute to the understanding of the TBVs community and also reveal the potential risks from TBVs being vectored by R. microplus ticks in Yunnan Province, China.
-
From 2015 to 2017, 7387 live adult ticks were plucked from infested cattle and buffalo in Nujiang Lisu Autonomous Prefecture (NJ), Dali Bai Autonomous Prefecture (DL) and Luxi County (LX) in Yunnan Province (Supplementary Figure S1 and Table 1), maintained alive in cases at room temperature before study. The ticks were identified morphologically as R. microplus by a trained expert and further confirmed by sequencing internal transcribed spacer 2 gene (ITS2). These ticks were washed three times using sterile phosphate-buffered saline (PBS, pH7.4), grouped (n = 11–20) depending on their body size and homogenized with 1 mL PBS for each group as described (Zhang et al. 2018). Homogenates were centrifuged at 5000 ×g for 10 min at 4 ℃ to remove debris, and supernatants were subpackaged and stored at - 80 ℃ until further processing.
Time Sampling location Number of groups Total ticks 2015 NJ 76 912 LX 45 540 2016 DL 351 4212 LX 47 564 2017 LX 79 410 Total 574 7387 Table 1. Summary of total tick sample collections in Yunnan Province from 2015 to 2017
Additionally, 100 serum samples of cattles, 100 serum samples from febrile patients and 100 healthy human serum samples from the same tick sampling location in LX were shipped to the laboratory in a cooler with ice pack or with dry ice, depending on the time required for transportation. Besides, 100 cattle serum samples were collected from a farm of Wuhan, Hubei Province.
-
According to our sampling location and time, our tick sample can be classified to four batches. And each 12 ticks from the same batch were divided into one group. For RNA-seq, three groups of ticks were mixed for one RNAseq library construction, and totally four libraries corresponding to each batches were constructed. Total RNA was purified using TRIzol LS reagent (Invitrogen, Calrsbad, CA) from 300 μL supernatant of each group. Then equivalent qualities of total RNA from three groups were mixed to generate one RNA pool (3–5 μg in total). RNA-seq library preparation was performed according to the protocol provided by Illumina as described (Li et al. 2015), and was further sequenced by the Hiseq 3000 platform (Illumina) to generate the 150 bp paired-end reads.
Raw sequencing reads with low quality were excluded, trimmed to remove adaptors using Trimmotic program and then subjected to a basic local alignment search tool (BLASTn) comparing against the tick genome to filter out the abundant host reads so as to simplify the following de novo assembly using Trinity program (Grabherr et al. 2011). Subsequently, the contigs were mapped to non-redundant nucleotide database (NT) and non-redundant protein database (NR) downloaded from NCBI using BLASTN and BLASTX respectively by using an E-value cutoff of 1e-5 to identify potential viral contigs. An overview of contig taxonomy was shown by MEGAN software with the BLASTN or BLASTX comparison results (Huson et al. 2007). Bowtie2 were used to obtain the raw reads of contig which mapping to a certain target viral genome and corresponding reads per kilobase million mapped reads (RPKM) were calculated (Langmead and Salzberg 2012).
-
Nested RT-PCR was performed to confirm the detection of RNA-seq with primer sets (Supplementary Table S1). That is cDNA from the tick pools which was originally subjected to RNA-seq were used as templates for PCR detection. Only positively detected potential virus would undergo further epidemiological survey.
The contigs which hit viral genome were used as templates for overlapping primers design. And the full-length genome of each virus was determined via genome walking, and 50- and 30- rapid amplification of cDNA ends (50-, 30 RACE) with commercial kit (Takara) according to the manufacturer's protocol.
All the PCR products were separated on 0.8% agarose gel and purified through agarose gel DNA extraction kit for Sanger sequencing.
-
Firstly, plasmid pET-28a-NP containing partial or complete nucleoprotein of each virus was constructed. Then recombinant plasmid pET-28-NP was electrotransformed into E.coli strain BL21 for protein expression induced with IPTG. And expressed NP was purified as described previously (Zou et al. 2016), which subsequently used for rabbit immunization to elicit polyclonal antibodies (pAbs) against NP. Western blots were performed to verify the effectiveness of the polyclonal antibody (anti-NP).
The same expression regions of pET-28a-NP were inserted into plasmid pCNDA3.1+ for eukaryotic expression. Then immunofluorescence assay was performed by using plasmid-transfected HEK293 cells. Cells were fixed with 4% paraformaldehyde for 15 min before being permeabilized with 0.5% Triton X-100 in PBS solution for 10 min. After rinsing with PBS solution, they were blocked with fish gelatin for 1 h, and then washed with PBS solution for another 3 min. The washing steps were repeated three times. Subsequently, bovine sera (diluted 1:20 in PBS solution) or polyclonal antibodies to NP (1:2000) as positive control group were incubated with cells for 2 h and washed as described earlier. Reactions among bovine sera were detected with FITC conjugated protein G (1:2000; Abcam) and the positive controls were detected with Alexa 488-labeled goat anti-rabbit (1:2000; Abcam). Cell transfected with empty pCDNA3.1+ was severed as negative control.
-
For each potential virus, phylogenetic trees were inferred based on the full-length RNA-dependent RNA polymerase (RdRp) protein which were aligned with their corresponding homologous references sequence. Alignment and phylogenetic analysis were conducted via ClustalW and maximum likelihood (ML) method which both implemented within MAGA version 6.0 (Tamura et al. 2013).
-
We use ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder) to predict potential protein, TMHMM v2.0 (https://www.cbs.dtu.dk/services/TMHMM/) to predict transmembrane domains, SignalP v4.0 (https://www.cbs.dtu.dk/services/SignalP/) to find likely signal sequence, NetOGlyc (https://www.cbs.dtu.dk/services/NetOGlyc/) to predict O-GalNAc glycosylation sites and NetNGlyc (https://www.cbs.dtu.dk/services/NetNGlyc/) to predict N-glycosylation sites.
Sample Collection and Preparation
Library Preparation and RNA-seq Sequencing
Viral Full-Length Genome Walking and PCRBased Epidemiological Survey
Preparation of Polyclonal Antibodies and Immunofluorescence Assay
Phylogenetic and Bioinformatics Analysis
Prediction of Protein Functional Domain
-
From 2015 to 2017, a total of 7387 ticks were collected from cattle and buffalo in the northwest (NJ and DL) and southeast (LX) areas of Yunnan Province and were divided into 574 groups (Supplementary Figure S1 and Table 1). Except that samples were collected continuously from LX for 3 years, ticks were collected from NJ in 2015 and from DL in 2016 (Table 1). These ticks were all half or full engorged R. microplus identified both by morphology and ITS2 sequencing (Fig. 1).
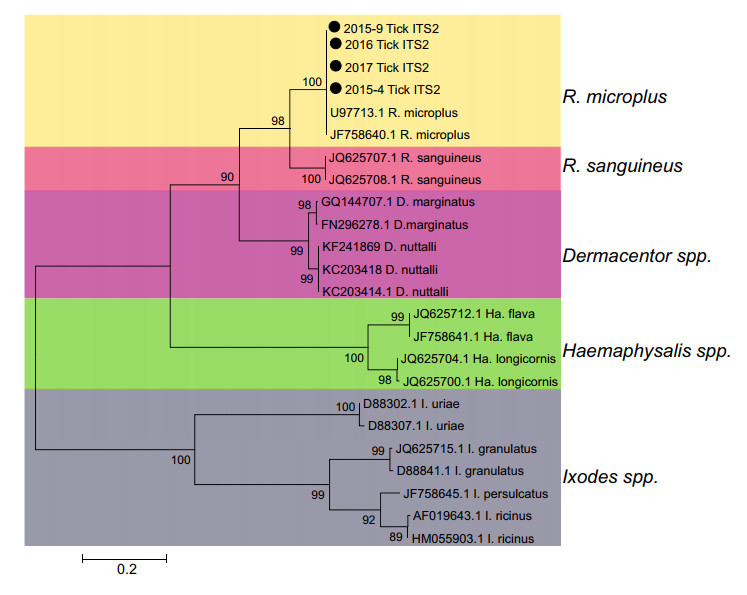
Figure 1. Molecular identification of tick samples. The phylogenetic analysis was performed via an ML method based on internal transcribed spacer 2 (ITS2) gene. The ticks collected in different year in this study were indicated by black dots.
Additionally, 100 serum samples of cattle and febrile patient and healthy human, respectively from Luxing County and 100 serum samples of cattle from Wuhan City in Hubei Province were acquired.
-
A total of four tick pools collecting from different regions of Yunnan during 2015 to 2017 generated ~ 28 Gb of data including 2.7 × 109 or so 150-base pair-end reads (Supplementary Table S2). After reads assembly and sequence comparison to the NT or NR database, 143 contigs from the four libraries were identified as viral sequences related to 9 viruses, concerning Phenuiviridae (n = 3), Chuviridae (n = 2), Rhabdoviridae (n = 1), Parvoriridae (n = 1) and Flaviviridae-like (n = 2) (Table 2).
No. Virus name Closest relative Identity with relative virus (aa) Classification of relative virus Coverage (%) RPKM 1 Yunnan mivirus 1 (YNMV1) strain YNMV1 Wuhan mivirus strain X78-1 86% Mivirus 94.2 4.8 2 Wuhan tick vrius 1 (WHTV1) strain YNrhabdoV1 Wuhan tick vrius 1 strain X78-2 97% Unassigned Rhabdovirdae 83.3 2.5 3 YN tick-associated phlebovirus 1 (YNPhelobV1) strain YN-PLV1 Lihan tick virus strain LH-1 S segment 89%
L segment 88%Phlebovirus 86 1.57
1.94 Jingmen tick virus (JMTV) strain YNflaviV Jingmen tick virus isolate SY84 S1 segment 93%
S2 segment 99%
S3 segment 96%
S4 segment 99%Unssigned (Jingmenvirus) S1:87.7
S2:84.5
S3:86
S4:87.21.33
2.7
1.8
3.05 Bovine hokovirus 2 (BHKV) strain YNBHKV2 Bovine hokovirus 2 strain HK-B38 100% Tetraparvovirus 97 1.30 6 Bole mivirus (BLMV) strain YN-MV2 Bole tick virus 3 strain BL199 95% Mivirus 44.1 0.09 7 Bole tick virus 1 (BLTV1) strain YNPLV2 Bole tick virus 1 strain BL075 S/L segment 98% Phlebovirus 36.4 0.05 8 YN tick-associated flavi-like virus 1 (YN-FlaviV1) strain YN-FLV 1 Bole tick virus 4 80% Unassigned (Flavi-like) 30.2 0.03 9 YN tick-associated phlebovirus (YNPhelobV1) strain YN-PLV3 Blacklegged tick virus S segment 60%
L segment 65%Phlebovirus 23.2 0.01
0.01RPKM: Reads Per Kilobase per Million mapped reads Table 2. Viruses identified in R. microplus in our study
RPKM values of the nine viruses suggested that the first five viruses (RPKM > 1.0) had much higher abundance than the last four viruses (RPKM < 1.0) (Table 2), in accordance with the genome coverage of RNA-seq that the former five viruses had several gaps along their genome sequences while dispersed short contigs flanked by large gaps were found along genome of the latter viruses. Genome walking was further performed with the first five viruses in order to acquire the full-length genome. Genome organization of these viruses was shown in Fig. 2, reflecting a great genomic diversity of tick-associated viruses.
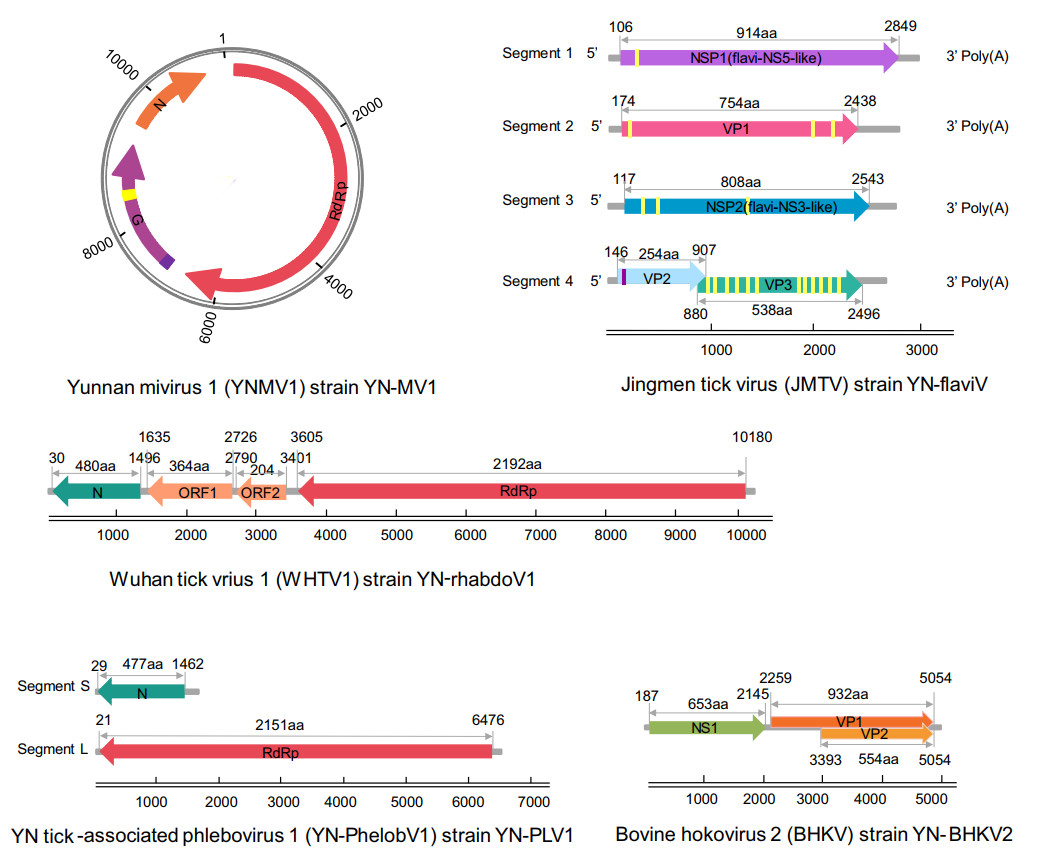
Figure 2. Schematic genome organization and putative proteomic maps of each virus. Yellow boxes represent the transmembrane regions (TM), and purple boxes represent the signal peptides (SP).
-
Viral sequences share 86% and 95% identity at amino acid (aa) level with Wuhan mivirus (WHMV) strain X78-1 and Bole mivirus (BLMV) strain BL199, respectively were detected in our tick pools. WHMV and BLMV are identified in the R. microplus and Hyalomma asiaticum tick in China, respectively and assigned to the recently-proposed Mivirus genus of Chuviridae (Li et al. 2015). Based on the similarity, we considered virus related to WHMV as a novel chuvirus named Yunnan mivirus strain YN-MV1, and virus related to BLMV as a novel strain of BLTV named YN-MV2. Here we obtained the full-length genome of YN-MV1 (Fig. 2). YN-MV1 (GenBank MH814982) is a negative-strand circular RNA virus similar to WHMV, with genome length of 11, 395 nt which contains three open reading frames (ORFs) encoding RNA-dependent RNA polymerase (RdRp), glycoprotein (G) and nucleoprotein (NP) (Fig. 2). Phylogenetic tree showed that YN-MV1 is a novel member of Mivirus (Fig. 3A).
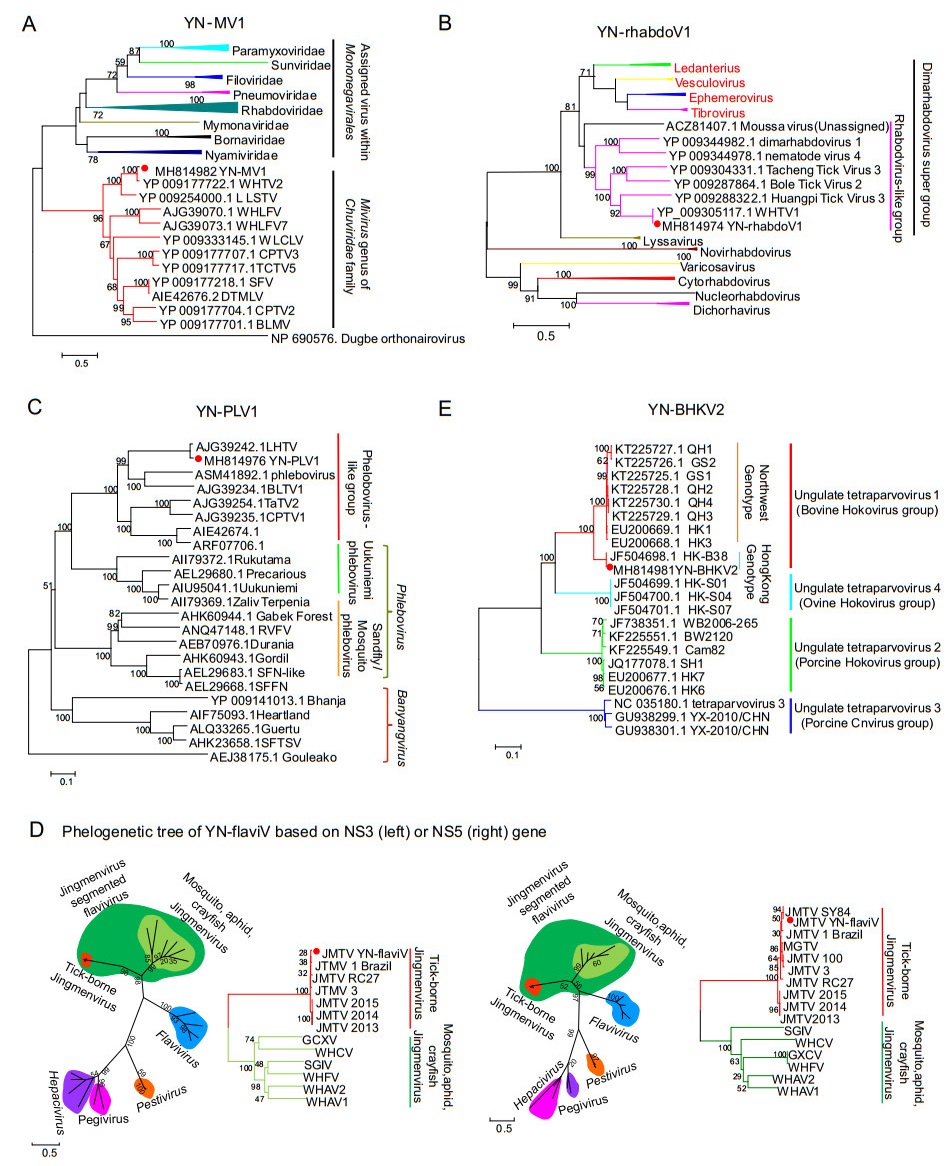
Figure 3. Phylogenetic relationship of A YNMV1 based on RdRp of members within Mononegavirales, B YN-rhabdov1 based on RdRp of rhabdovirus, C YN-PhleboV1 based on RdRp of bunyavirus, D YNflaviV1 based on NS3 and NS5 of flavivirus, E YN-BHKV2 virus based on RdRp of tetraparvovirus (indicated by red dot) with their closest viral family or genus. The trees shown here were inferred using an ML method with 1000 bootstraps.
-
Via blastn search, viral sequences demonstrated 97% identity to the Wuhan tick virus 1 (WHTV1) strain X78-2 were found in our tick pools. It is proposed to be a strain of WHTV1 termed YN-rhabdoV1 (GenBank MH814974). WHTV1, originally detected in same ticks in Hubei Province, currently remain unclassified in the family Rhabdoviridae and considered as a member of the dimarhabdovirus super group (Bourhy et al. 2005). The single negative-strand genome of YN-rhabdoV1 with a 10, 306 nt in length consists of four ORFs, two of which encodes RdRp and NP both being homologous with that of rhabdovirus (Fig. 2). The functions of the remaining two ORFs (ORF1 and ORF2) are unknown for no homologous known protein has been found. Phylogenetic analysis with other rhabdovirus shows that YN-rhabdoV1 is closer to Moussa virus, which is an unassigned rhabovirus, than to the other genus and forms a separated branch with a group of novel rhabodovirus-like virus identified mainly in ticks via NGS (Fig. 3B). This group of novel rhabodovirus-like virus may be considered as a new genus within Rhabdovirdae.
-
A number of tick-associated phlebovirus sequences were identified in our tick pools, and blastn or blastx search indicate these sequences mostly relate to viruses of Phlebovirus genus in Phenuiviridae. Viral sequences share 89% aa identity with Lihan tick virus (LHTV) which was originally identified in same tick species in central China, were considered to belong a novel phlebovirus named Yunnan tick-associated phlebovirus 1 strain YN-PheloboV1. Sequences share 98% aa identity with Bole tick virus 1 (BLTV1) strain BL075 are supposed to belong to a novel strain of BLTV1, tentatively named BLTV1 strain YNPheloboV2. And the remaining short contigs are considered as a novel phlebovirus named Yunnan tick-associated phlebovirus 3, sharing 60% aa identity with a blacklegged tick virus, which was originally identified in blacklegged tick from America. Like other phlebovirus, the genome of YN-PheloboV1 is supposed to consist three negative RNA segments termed S, M and L, respectively (Fig. 2). However, the M segment of both YN-PheloboV1 and LHTV haven't been identified yet. Phylogenetic tree shows that YN-PheloboV1 (Genbank No. MH814975 for S, Genbank No. MH814976 for M) is closest to Uukuniemi virus forming a separated branch with a group of novel putative phleboviruses which are characterized with unknown M segment (Fig. 3C).
-
Recently, a group of four segmented single-stranded positive RNA viruses related to flavivirus were identified in various arthropod and the prototype virus Jingmen tick virus (JMTV) strain SY84 was the firstly identified in R. microplus ticks collected in Hubei Province, China (Shi et al. 2016). Here, we also found the genomic sequences of a new JMTV strain YN-flaviV in the R. microplus ticks collected in 2017 in LX of Yunnan Province, which showed 93%–99% nt identity with strain SY84. Like the genome of JMTV, YN-flaviV comprises four segments (GenBank MH814977-MH814980) (Fig. 2), among which segment 1 and segment 3 respectively code protein related to nonstructural protein NS3 and NS5 of Flavivirus (family Flaviviridae). Proteins coded by segment 2 and segment 4 are quite unknown because no homologous proteins have been found. Via bioinformatics prediction, two N-glycosylation sites and three transmembrane domains were found on VP1 coded by segment 2, indicating VP1 may be a glycoprotein. Segment 4 has two ORFs coding VP2 and VP3. One signal peptide on the N terminal, one N- and O-glycosylation site was predicted on VP2. And VP3 is largely a membrane protein because 11 transmembrane domains were predicted on it. Phylogenetic tree with members of Flaviviridae shows that YN-flaviV clusters with a group of four segmented flavirus-like viruses named Jingmenvirus group, and were closest to members of Flavivirus (Fig. 3D). And this highly divergent lineage obviously separates into two sub-clades with one related to ticks and the other one related to insects or arthropods. We also found some short contigs related to NS3 and NS5 protein of flavivirus which share 80% aa identity with Bole tick virus 4 (BLTV4). BLTV4 was originally identified in Hyalomma asiaticum in Xinjiang Province of China and remained unclassified (Shi et al. 2016). We consider these flavi-like sequences detected in our study belong to a novel virus named Yunnan tick-associated flavi-like virus 1 (YNFLV1).
-
A new strain of Bovine hokovirus 2 (BHKV2) was found in our 2017-YNT RNA-seq library named YN-BHKV2 (GenBank MH814981), the genome of which shares 100% identity with BHKV2 strain BS-S13, which belongs to Tetraparvovirus genus of Parvoviridae. The genome of YN-BHKV2 consists of two ORFs, with ORF1 encoding the non-structural protein (NS1) and ORF2 encoding the vial capsid protein (VP1) as well as a putative transmembrane protein (VP2) encoded by an overlapping reading frame in ORF2 (Fig. 2). Phylogenetic analysis with other viruses of Tetraparvovirus genus shows YN-BHKV2 clustered with bovine hokovirus within ungulate tetraparvovirus group 1. This group of viruses was mainly detected in bovine from Hong Kong and domestic yaks in northwest China. However, different from the genotype epidemic in northwestern of China, another genotype is epidemic in Yunnan Province which was identical to that of Hong Kong (Fig. 3E).
-
PCR-based and immunofluorescence-based epidemiological survey was performed to investigate the prevalence of each virus in natural R. microplus tick during 2015–2017 (Table 3) and cattle and human sera (Table 4 and Fig. 4). IFA analysis of the positive control shows that the NP of each virus can be effectively expressed via transfected HEK293 (Fig. 4). YNMV1 virus has 20% or so positive rate among ticks from different landforms in different year. As for WHTV1, significantly high positive rate (42%) was detected in NJ in 2015, and relatively low positive rate (5%) or so was detected in LX in 2016 and 2017. Remarkably, serological assay shows high positive rate as 89% and 94% for YNMV1 and WHTV1, respectively among cattle serum. Comparing the above two viruses, prevalence of YN-PhleboV1 is much lower with 2.6% to 18.4%. Consistently, 8% cattle serum was positively detected with anti-YN-PhleboV1 antibody. And JMTV was only positively detected in one group of ticks from LX collected in 2015. Serological survey shows 18% cattle serum samples were detected positive for JMTV, meanwhile RNA was detected in four of these seropositive samples. Additionally, 63.1% tick samples from LX in 2017 and 40% cattle serum samples are positive for BHKV2. We also performed the same serological survey on 100 bovine serum samples from Hubei Province and healthy/febrile human serum from Yunnan LX, and no positive sample was detected with these five viruses. Collectively, these results suggest a stable and wild existence of YNMV1, WHTV1 and YN-PhelobV1 in R. microplus of Yunnan Province via circulation among ticks and cattle or may be other mammals.
2015 2016 2017 NJ
(76)LX
(45)DL
(353)LX
(41)LX
(79)Yunnan mivirus 1 28.9%
(22/76)24.4%
(11/45)24.9%
(88/353)19.5%
(8/41)17.8%
(14/79)Wuhan tick vrius 1 42%
(32/76)12.9%
(4/45)15.8%
(56/353)4.8%
(2/41)5%
(4/79)YN tick-associated phlebovirus 1 2.6%
(3/76)11.1%
(5/45)18.4%
(7/31)4.8%
(2/41)15%
(12/79)Jingmen tick virus 0 1/45
(2.2%)0 0 0 Bovine hokovirus 2 0 0 0 0 63.3%
(50/79)Table 3. PCR-based epidemiological survey of each virus among tick samples
Cattle Serum sample Human serum sample Hubei
(n = 100)Yunnan
(n = 100)Febrile patient
(n = 100)Healthy human
(n = 100)Yunnan mivirus 1 0 89% (89/100) 0 0 Wuhan tick vrius 1 0 94% (94/100) 0 0 YN tick-associated phlebovirus 1 0 4% (4/100) 0 0 Jingmen tick virus 0 18% (18/100) 0 0 Bovine hokovirus 2 0 40% (40/100) 0 0 Table 4. IFA-based epidemiological survey targeting NP protein of each virus
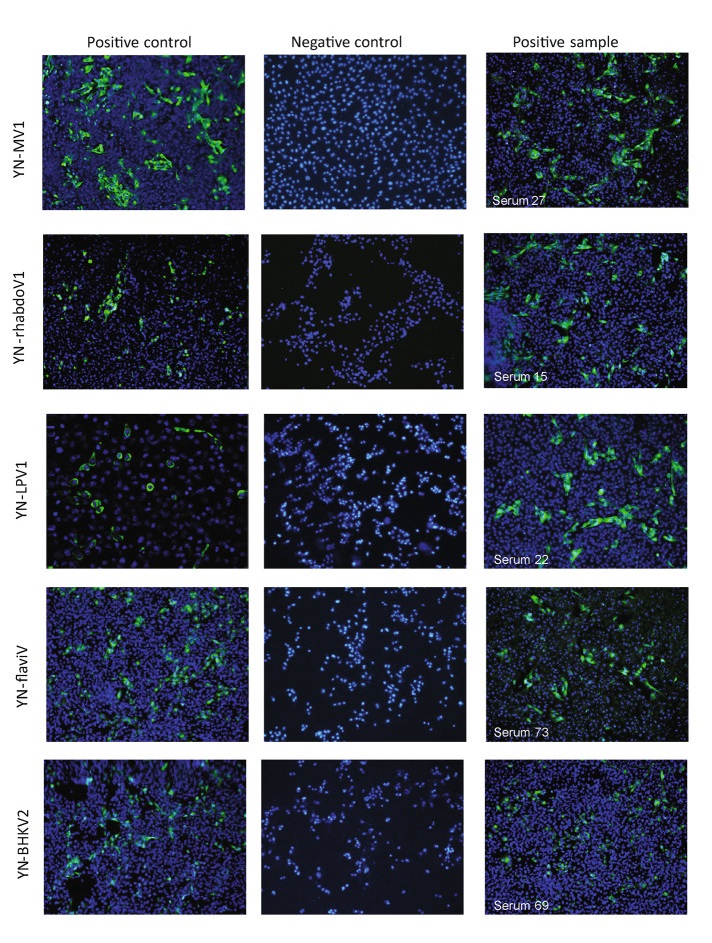
Figure 4. Representative results of seropositive cattle serum samples detected via IFA. Polyclonal antibody to NP was the primary antibody in positive control, and HEK293 transfected with empty pCDNA3.1+ plasmid in negative control. All the photos were taken at ×20 magnification.
Tick Sample Collection and Taxonomic Identification
Metagenomic Sequencing Revealed Virome Profile of R. microplus Ticks
Viruses Belong to Chuviridae
Viruses Belong to Rhabdoviridae
Viruses Belong to Phenuiviridae
Flavi-like Viruses and Jingmenvirus
Viruses Belong to Parvoviridae
Virus Prevalence Among R. microplus Tick and Serum Samples
-
Our primary objective is to throw light on the virome profile of primary ticks R. microplus in Yunnan Province, which is the most important ectoparasite and disease vector of livestock globally. The most notorious pathogens transmitted by R. microplus are Babesia bigemina, B. bovis and Anaplasma marginale (Parizi et al. 2015), which cause large economic loss in livestock industry. Comparing to the pathogenic parasites, fewer viruses related to R. microplus were reported. Investigating the viral profile of R. microplus will improve our understanding on the viral pathogens of R. microplus. Via three consecutive years of metagenomic sequencing, totally nine viruses concerning chu-, rhabdo-, phlebo- and flavilike viruses were identified indicating the circulation of diverse tick-associated viruses in Yunnan.
The study identified two chuviruses with on novel species (YN-MV1) and one strain of a previously reported Bole mivirus (YN-MV2). And complete genome of YNMV1 and partial sequence of YN-MV2 were obtained. Chuviruses constitute a large monophyletic group, locating at an intermediated phylogenetic branch between segment and unsegmented negative-sense RNA viruses. Members of chuvirus characterized with diverse genome organization including unsegmented, bi-segmented and circular forms are commonly identified in arthropods like ticks, spiders, true flies and crabs (Li et al. 2015; Brinkmann et al. 2018). Here, we complementarily provide the evidence that chuvirus is ubiquitously prevalent among R. microplus ticks in Yunnan, southwest of China.
An increasing number of unassigned rhabdoviruses including the WHTV1 are identified within various arthropods (Li et al. 2015; Brinkmann et al. 2018). Because only proteins of RdRp and NP similar with that of rhabodovirus could be identified while the remaining corresponding proteins are absent in these group of rhabdovirus, which made it difficult to define their taxon within Rhabdoviridae. And it is proposed that these viruses should be considered as a member of the dimarhabdovirus supergroup (Bourhy et al. 2005; Brinkmann et al. 2018) (Fig. 3B). Our viral sequence findings further contribute to the understanding of viral taxon definition. And PCR-based survey for YNMV1 and WHTV1 among ticks provides preliminary evidence for their widespread circulation in R. microplus.
Three distinct tick-associated phleboviruses were detected with two novel phleboviruses (YN-PhelobV1, YN-PhelobV3) and one variant (YN-PLV2) of prototype virus Bole tick virus 1. And nearly complete S and L segment of YN-PLV1 were characterized while only short contigs of YN-PLV2 and YN-PLV3 were found. Phylogenetic analysis consistent with sequence comparison demonstrates YN- PLV1 mostly related to Uukuniemi phlebovirus and clustering with a group of novel phleboviruses which characterized with unknown M segment presently (Fig. 3C). The great diversity of glycoprotein coded by M segment make it difficult to search homologous protein for this group of novel viruses via homologous sequence alignment. Or, we can't exclude the possibility of the existence of bi-segment viruses which resemble to Phlebovirus, because examples of fully viable two-segmented Rift Valley fever virus (RVFV) (Phenuiisridae, Phlebovirus) variant lacking the M-segment were documented (Kortekaas et al. 2011; Wichgers Schreur and Kortekaas 2016). We are working on virus isolation and viral particles rescue via reverse genetic system, hoping to throw light on these confused puzzles.
Consecutive positive detection of YNMV1, WHTV and YN-PhleboV1 in ticks strongly indicates a stable and widespread circulation of these viruses among R.micrplus in Yunnan. And all the ticks in our study are engorged and plucked from infested cattle, along with the significant high seropositive rate of 89% and 94% of YNMV1 and WHTV1 respectively. It's reasonable to speculate that these viruses are circulated via ticks among reservoir host including cattle or even other mammals, which deserve further research. Therefore, all the identified viruses may have the ability to infect cattle and elicit corresponding antibody. However, the lack of observed disease in cattle infected with identified viruses may be a consequence of longestablished and stable co-existence of virus and host. But the possibility of changing vector or host adaptation causing by virus innate evolution should be taken into consideration. It would therefore be pertinent to initiate monitoring for the spread of these viruses to other animal species and new geographical locations.
Remarkably sequences related to parvovirus were only detected in ticks collected in LX in 2017. And the fulllength genome of our strain YN-BHKV2 was identical to the prototype Bovine hokovirus 2 (BHKV2) strain HK-B38 which identified in beef from Hong Kong meat market and belonged to Ungulate tetraparvovirus 1 group (Cibulski et al. 2016). BHKV2 is a well confirmed infectious agent to cattle, we speculate that the positive tick groups of YNBHKV2 in 2017 originate from infected domestic cattle, which can also be reflected by the high seropositive rate of 40% among cattle sera. And BHKV2 previously was reported broadly distributed in domestic yaks in northwestern China (Xu et al. 2016). Here, we firstly reported BHKV2 circulation in ticks of Yunnan indicating a wider circulation zone. What's more, BHKV2 belongs to the subfamily Parvovirinae whose members infect vertebrates while members of subfamily Densovirinae infect insets and arthropods (Cotmore et al. 2014). However, our findings suggested the probability that members of Parvovirinae may infect ticks and even transmitted via it, which deserves further investigation.
Finally, we have identified viral sequences related to flavivirus, which demonstrated 28% or so similarity to NS3 and NS5 protein of flavivirus. Sequences share 80% identity with BLTV4, which originally identified in Xinjiang Province of China in Hyalomma asiaticum, and it was proposed as a novel flavivirus-like virus named as YNFlaviV1. Previously study shows a number of flavi-like viruses are detected via metagenomics in a range of arthropod species, including ticks, true flies, mosquitoes and spiders (Brinkmann et al. 2018). Though failed to obtain the full-length genome of YN-FLV1, our findings supplementarily suggest that tick-associated flavivirus-like viruses are common and ubiquitous in R. microplus of Yunnan. The remaining flavi-related sequences show 93%- 99% identity with JMTV strain SY84, which is a novel discovered four segmented RNA virus with phylogenetic relationship with flavivirus. Nearly complete genome of JMTV strain YN-flaviV was obtained in our study. Though only one group of ticks from LX in 2015 was detected with YN-flaviV, 18% seropositive results indicate this flavi-related virus may be circulated in Yunnan especially among cattle. And RNA of YN-flaviV was detected in four of the seropositive serum samples which further confirmed the serological result based on IFA. Previous prevalence survey indicated other ticks including Ixodes spp., Armigeres spp. and Haemaphysalis spp. from China and R. microplus from Brazil also carry JMTV (Qin et al. 2014; Souza et al. 2018). And it was subsequently found in the plasma of a red colobus monkey in Uganda (Maruyama et al. 2014). In addition, scientists have detected full-length genome of JMTV in three human Crimean-Congo hemorrhagic fever cases though no obvious symptoms were observed (Emmerich et al. 2018). And another Jingmenvirus member named Alongshan virus (ALSV) has more direct evidence for infectious to human since it is isolated from serum of a fever patient (Wang et al. 2019). And a number of reads with high similarity with Jingmenvirus have been identified in wild P. leucopus mice from North America (Vandegrift et al. 2020). Combined with our findings, it's reasonable to speculate that JMTV can circulate among domestic cattle and other unidentified vertebrate animals vectored by ticks, especially R. microplus. And questions about potential pathogenic effect of JMTV on human and animal health deserve further attention.
Many members of Flavivirus are TBVs and pathogenic to human or animals, such as tick-borne encephalitis virus (TBEV), Kyasanur Forest disease virus (KFDV), Louping ill virus (LIV). So, it is important to investigate this virus in respect of both evolution and pathogenicity to public health. We have tried to isolate JMTV via inoculation on new-born mice and kinds of mammal cell lines, including HEK293, BHK-21, Vero, PK15 and DH82 cell lines but failed. Recently, researchers have reported the successful isolation of JMTV via tick cell line BME/CTVM23 or BME26 and Vero cells, however, these cells seem not suitable enough for JMTV consistent maintenance. Very low virus titer and lack of plaque greatly hinder further study on JMTV (Dinçer et al. 2019; Jia et al. 2019). The investigation on the optimal cell line for JMTV and other Jingmenvirus propagation remains to be resolved. And to obtain in-depth understanding of transmission cycle on this flavi-related virus, further epidemiological survey including other domestic animals and ticks should be considered. Furthermore, the existence of JMTV in monkey, cattle and febrile patient should be a warning that it may be a potential emerging zoonoses and/or tick-borne virus.
Notably, no positive sample was detected in serum samples from Hubei Province. However, JMTV1 and WHTV1 were all originally identified in R. microplus tick in Hubei Province (Li et al. 2015). We speculate that it is because the serum samples of Hubei come from the same farm while serum samples of Yunnan from scattering cattle raising households, which are more possible to expose to ticks. Such difference appears to reflect the role of ticks in transmitting viruses. Further surveillance on these viruses in different regions will contribute to the information of their geographical distribution.
Summarily, we provide a comprehensive investigation on virome profiles in R. microplus tick in Yunnan. Though no known viruses causing disease to human and animals were detected, our results can confirm the presence of chu-, rhabdo-, phlebo-, flavi-like and parvovirus in R. microplus in Yunnan. And epidemiological survey suggests the stable and widespread circulation of YNMV1, WHTV1 and YN-PhleboV1 among ticks and cattle. Meanwhile, the occasionally positive tick and moderate seropositive cattle serum samples of segmented flavi-related virus and the parvovirus indicating the parasitized vertebrates may be the source in some instances. Our work highlights a remarkable diversity of RNA virus community and extends the current viral diversity. However, further studies are needed to reveal the role of these viruses in the pathogenesis of animals and humans.
-
This work was jointly funded by the Scientific and Technological Basis Special Project grant (2013FY113500) from the Ministry of Science and Technology of China, the National Natural Science Foundation of China (81874274 and 81660558), the National Science and Technology Major Project on Important Infectious Diseases Prevention and Control (2018ZX10734-404), and the Yunnan Health Training Project of High Level Talents (L-2017027).
-
SS, YZ and FD conceived and designed the experiments. JS, and HW performed the experiments. JS, SS and FD wrote the paper. YZ and FD finalized the manuscript. All authors have read and approved the final manuscript.
-
The authors declare that they have no conflicts of interest.
-
Collections of ticks and serum of cattles were approved by the Medical Ethics Committee of Dali University. Human serum samples were collected from patients and healthy volunteers with informed consent. All the procedures performed in studies involving animals were in accordance with the ethical standards of the Animal Care and Ethics Committees of Wuhan institute of virology, CAS (Approval numbers WIVA01201601).

 DownLoad:
DownLoad: