












HTML
-
Influenza virus is a negative-sense single stranded RNA virus which belongs to the family of viruses known as Orthomyxoviridae. According to the ICTV (https://talk.ictvonline.org/taxonomy/), there are four types of influenza viruses: A, B, C and D. Human can be infected only with influenza A, B and C viruses, while influenza D virus primarily affects cattle (Su et al. 2017). Influenza A and B viruses are responsible for the worldwide pandemics and the seasonal epidemics which can cause tremendous loss of human lives and social economy. The World Health Organization (WHO) estimated that approximately 3 to 5 million cases of severe illness resulted from seasonal influenza epidemics, and that about 290, 000 to 650, 000 people died of the respiratory illness worldwide (https://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal)).
-
As influenza A and B viruses are the primary threats to humanity, a lot of studies have been done on these two viruses. The genomes of influenza A and B viruses both consist of eight RNA negative-sense segments, including six internal genes (PB1, PB2, PA, NP, M, NS) and two external genes (HA, NA), which encode more than ten proteins in total. The segmented nature of the viral genome will lead different viruses to exchange the intact gene segments when they co-infect the same host cell. This important evolutionary mechanism of influenza viruses is called reassortment (Fig. 1). Novel viruses with new characteristics can be created by reassortment, which may have great pandemic potential in humans. In fact, all the viruses with multiple genomic segments may reassort, like bunyaviruses and reoviruses. However, most reports and researches of the genomic reassortment are about influenza virus (Blitvich et al. 2012; Ahasan et al. 2019). Among the four previous pandemics, three have been ascertained to be associated with reassortments. The 1957 Asia Flu pan-demic was triggered by the new H2N2 subtype, a reas-sortant between an avian influenza virus and the H1N1 virus from the previous 1918 pandemic (Kawaoka et al. 1989; Schäfer et al. 1993; Kilbourne 2006; Smith et al. 2009b). Later, the 1968 Hong Kong Flu pandemic was caused by the H3N2 reassortant which combined six genes (PB2, PA, NA, NP, NS and M) of the H2N2 virus from the 1957 pandemic and two genes (PB1 and HA) from a novel avian influenza virus (Kawaoka et al. 1989; Kilbourne 2006; Smith et al. 2009b). Recently, in the 2009 flu pan-demic, the novel H1N1 virus was a triple reassortant of an avian influenza virus source (the PB2 and PA genes), the human seasonal H3N2 influenza virus (the PB1 gene) and the swine influenza viruses (the HA, NP, NS, NA and M genes) (Garten et al. 2009; Smith et al. 2009a; Mena et al. 2016). In addition, influenza virus reassortants of some avian sources may spread to humans by overcoming a series of transmission barriers (Gao et al. 2013; Xing et al. 2016). For example, a novel H7N9 virus in 2013 that caused an endemic in China was created by the genomic combination of the original H7N9 virus and the H9N2 virus (Lam et al. 2013; Wu et al. 2013).
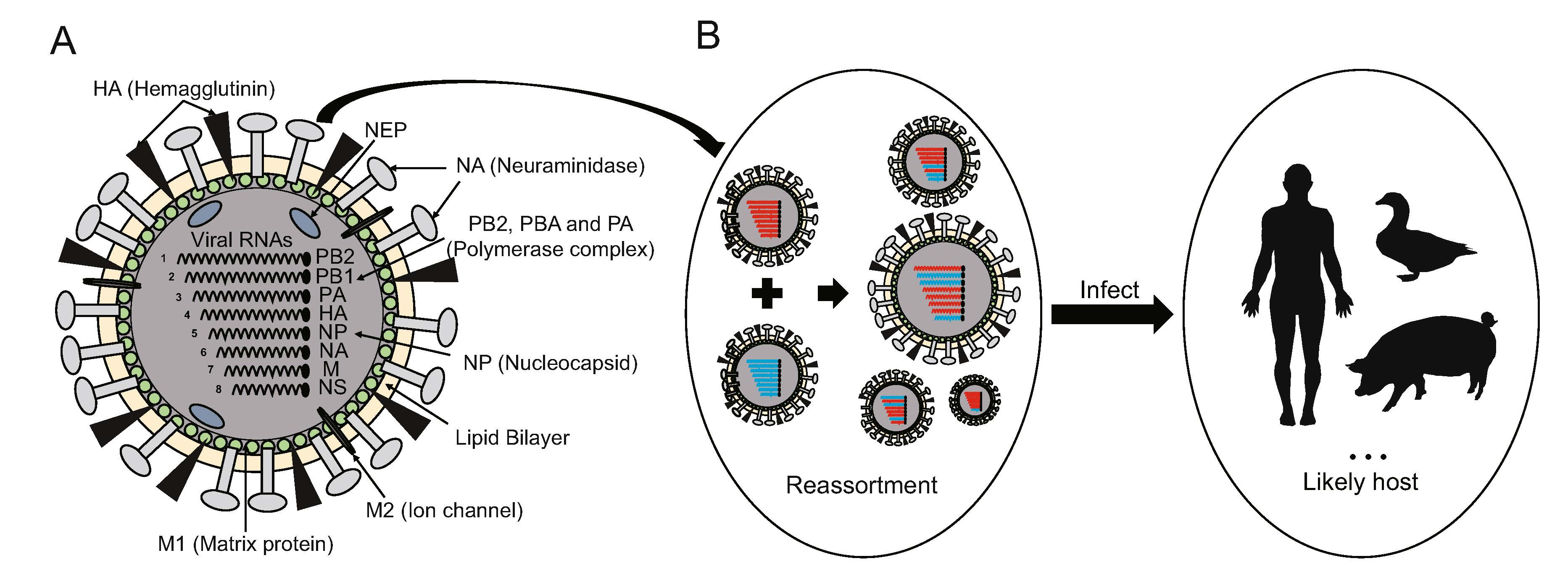
Figure 1. Genomic structure of influenza viruses and the principle of influenza virus reassortment. A The genome of influenza viruses contains eight RNA negative-sense segments including six internal genes (PB2, PB1, PA, NP, M and NS) and two external genes (HA and NA). More than ten proteins correlated with viral structure and function are encoded by these eight segments. B The principle of influenza virus reassortment. The reassortants can be produced by exchanging the gene segments when different influenza viruses co-infect the same host cell, which may cause influenza pandemics in different animals.
-
As the reassortment underlies the important evolution for influenza viruses, increasing experimental efforts were made to study the reassortments. However, such research is challenging due to the safety and ethical issues (Butler 2011; Fouchier 2015). With the advent of the big data era, more and more influenza virus genomic data are generated. Additionally, the improvements were made on bioinfor-matics by the development of various subjects like math-ematics, physics and biology, etc., leading the computational methods to be one of the indispensable tools for studying the influenza virus reassortment. As shown in Fig. 2, the key point of the data-driven computational identification of reassortments is to recognize the hetero-geneity of multiple gene segments based on the genomic data; then the integrative analyses with related epidemio-logic information are made to infer the reassortment events. Many tools that were designed to detect virus genomic recombination have been developed, which can also identify the virus reassortment, such as Simplot (Lole et al. 1999), Recombination Detection Program (RDP) (Martin and Rybicki 2000), GENECONV (Sawyer 1989), DSS (Difference of Sums of Squares) (McGuire et al. 1997), MAXCHI (Maximum Chi-Square method) (Smith 1992), and so on. Currently, the specific bioinformatics identification methods are mainly divided into two types as summarized in Table 1, the phylogenetic tree-based methods and the phylogenetic tree-independent methods. Here, a comprehensive review of influenza virus reassortment identification is given in this section. By summarizing the existing computational approaches in reassortment identification, we believe this work can serve as a reasonable guide to identify and infer reassortments.

Figure 2. The framework of reassortment identification with bioinfor-matics methods. Reassortants can be inferred by recognizing the heterogeneity of multiple segments based on the genomic data first, as shown in the left panel. Then the heterogeneity and related epidemiologic information, such as sampled region, host and sampled date will be combined to identify the reassortment events further.
Classification Method Principle Accessibility Compiling environment Using experience Required data Limitation References Phylogenetic tree-based FluReF A bottom-up research Source code Written by C ++ on Linux system The test dataset containing 1050 strains spent 10 s in total Complete genome of influenza virus High computational complexity Yurovsky and Moret (2011) Villa et al. Core mutations Source code Written by both C++ and python on Linux system The mock test dataset containing 7477 strains with the 290 bp simulated genomic sequence took more than 5 days in total Complete HA and NA sequences Limited to the reassortment identification of the HA and NA segments Villa and Lassig (2017) FluResort Identities of predicted protein Web invalid Written with ANSI/ISO standard C ++ on both Windows or Linux systems Not available Viral protein sequences and mass spectral data of these proteins Limited to the HA, NA, NP and M1 proteins, and the mass spectral data with high-resolution was required Lun et al. (2012) Nagarajan et al. Enumerating maximal bicliques Not supported Not supported Not available Genomic segments of influenza virus High computational complexity Nagarajan and Kingsford (2008) GiRaF Graph theory Source code Written by C ++ on Linux, Mac or Windows systems The test dataset containing 35 strains took about 5 s in total Complete genome of influenza virus High computational complexity Nagarajan and Kingsford (2011) Suzuki et al. Topologies of quartet trees Not supported Not supported Not available Complete genome of influenza virus High computational complexity Suzuki (2010) Dong et al. Genotype profile IVEE soft Written by both C ++ and python on Windows system Each complete genome took 3 about seconds Complete genome and the genotype information It had limitations when inferring intra-subtype reassortments within the same host Dong et al. 2011) Phylogenetic tree independent Wan et al. Network module; MST Not supported Not supported Not available Genomic segments of influenza virus Not suitable for short sequences Wan et al.(2007a, 2007b, 2008) Rabadan et al. Hamming distance Not supported Not supported Not available Genomic segments of influenza virus The assumption of equal mutation rate among segments may not always hold Rabadan et al. (2008) Silva et al. Genetic distance Not supported Written by Ruby script using bioruby on a Debian Linux server system Not available Genomic segments of influenza virus The performance of this algorithm will be influenced by the sample bias significantly de Silva et al. (2012) HoPER Host tropism Not supported Not supported Not available The full-length amino acid sequences of all genomic segments It is difficult to identify the reassortments between different hosts Yin et al. (2020) Table 1. A brief review of the reassortment identification methods for the influenza viruses.
-
Currently, the phylogenetic incongruences of the relation-ships among eight gene segments in influenza viruses were identified to infer the reassortment events manually in most previous studies (Arenas and Posada 2010; Boni et al. 2010). In this process, the phylogenetic trees for each gene segment were first constructed with different methods such as neighborhood joining (NJ) (Saitou and Nei 1987), maximum likelihood (ML) (Felsenstein 1981) and maxi-mum parsimony (MP) (Sourdis and Nei 1988) etc., or based on the molecular clock analysis (Takezaki et al. 1995); the reconstructed phylogenetic trees were then partitioned into multiple clades manually based on some criteria like the bootstrap of ancestral node and the diver-gence time for different phylogenetic clades etc.; finally, the reassortment events would be recognized with the integration of topological incongruence and related epi-demiologic information. Lots of achievements with this manual identification approach were made on different subtype influenza viruses. For example, our previous work combined the molecular clock analysis and the viruses' epidemiological data to demonstrate that at least two sequential reassortments of the novel H7N9 viruses were took place with the distinct H9N2 viruses. The computa-tional results indicated that the first reassortment likely occurred in wild birds while the second occurred in domestic birds in east China in early 2012 (Wu et al. 2013). Recently, the genetic origin and evolution of H5N6 viruses were explored also with this method by Bi et al. (2016). In their work, a comprehensive phylogenetic analysis of eight gene segments with ML method coupling with the epi-demiological data was performed, which revealed that H5N6 arose from the reassortment of H5 and H6N6 viru-ses, and that the internal genes were constantly reassorted among low-pathogenic avian influenza viruses. In addition, the reassortment events of pandemic H1N1/2009 virus were successfully identified in Vijaykrishna's (Vijaykr-ishna et al. 2010) and Smith's work (Smith et al. 2009a) with similar methods.
Despite the significant achievements, the feasibility and validity of this identification method was still limited by the manual operation. Particularly, enormous amounts of genomic data on influenza viruses made the manual reas-sortment identification not available. Hence, automatic comparison among phylogenetic trees based on different gene segments was developed to improve the algorithm feasibility. FluReF is a fully automated reassortment finder which was proposed by Yurovsky and Moret (2011). The reassortment events can be identified by a bottom-up search for candidate clades on both the whole genome-based and segment-based phylogenetic trees, which sepa-rates the phylogenetic clades containing the reassortants from the other clades. As demonstrated in this work, FluReF could find reassortments effectively even for geo-graphically and temporarily expanded datasets. Recently, Villa et al. successfully inferred the reassortment events with the self-defined core mutations in genealogical trees in the investigation of the fitness cost in human influenza virus reassortment (Villa and Lassig 2017). Apart from the epidemiologic and mutational information, the biophysical data were also used. Lun et al. proposed a set of automatic reassortment identification algorithms, FluShuffle and FluResort (Lun et al. 2012). In FluShuffle, PepGen was first employed to generate theoretical peptide monoisotopic masses based on the influenza viral protein sequences. Then a Bayesian Markov Chain Monte Carlo (MCMC) approach (van Ravenzwaaij et al. 2018) was implemented to assign a combination of protein accessions to a single mass spectrum. Next, a Gibbs sampling algorithm was employed to estimate the marginal posterior probability for each known protein accession. Finally, accessions that match more peaks or match uniquely to a peak were selected with a higher probability at each step in the Gibbs sampler. The different combinations of influenza viral protein identities had been established through FluShuffle, which were then mapped onto the phylogenetic trees using FluResort. A statistical model was developed in FluResort to calculate the likelihood of reassortments, which was quantified using Z-score, a standardized value of the weighted mean patristic distance of each identity across different trees. This set of algorithms were evaluated with both the experimental and simulated mass spectral data obtained from the whole virus digests. For the experimental data, the algorithms were first tested with mass spectral data obtained from the digestion of a H1N1 strain from the reassortment of a 2009 H1N1 pandemic strain (A/Cali-fornia/07/2009) and a lab-modified H1N1 strain (A/Puerto Rico/08/1934). The seasonal influenza A and B viruses were also analyzed with these two algorithms. In addition, FluShuffle and FluResort algorithms were tested with the simulated mass spectral data. As indicated in this paper, these two algorithms accurately identified the natural reassortment of the H1N1 vaccine strain with the identifi-cation of each viral protein. Additionally, no reassortment events were recognized in the seasonal strain analyses. Although this set of algorithms can identify the reassort-ments accurately and rapidly, the mass spectral data with high-resolution are required.
Additionally, the graph theory was employed when many efforts were made on the automatic comparison of the phylogenetic trees. A framework based on the enu-merating maximal bicliques was first proposed to detect the reassortment events by Nagarajan and Kingsford (2008). Then, a fully automatic reassortment identification algo-rithm, GiRaF (Graph-incompatibility based Reassortment Finder) (Nagarajan and Kingsford 2011), was developed on the basis of this framework. In GiRaF, large groups of Markov chain Monte Carlo (MCMC)-sampled trees are searched for incompatible splitting by a fast biclique enu-meration algorithm coupled with several statistical tests to identify the differential phylogenetic topology. Then, the reassortment events are recognized with the combination of the differential from multiple gene segments. Three influ-enza virus datasets, including 156 human influenza A (H3N2) isolates (Levin et al. 2005), 35 avian influenza A (H5N1) isolates (Salzberg et al. 2007) and 140 swine influenza isolates (Kingsford et al. 2009) were evaluated with GiRaF, which had been analyzed in previous studies relying on the manual reassortment identification method. Not only the known reassortment events in these three influenza virus populations were detected accurately, but also several unreported reassortments in H5N1 and swine influenza isolates were identified. In addition, GiRaF can identify the reassortment events with high sensitivity as well as high precision for the simulated reassortment datasets. Recently, the reassortment events within the Victoria and Yamagata lineages were recognized by GiRaF when researchers exploited the evolutionary trajectories of influenza B viruses (Virk et al. 2020). A method based on quartet trees was proposed by Suzuki to detect reassort-ments, which can be used even when the constructed phylogenetic trees were unreliable (Suzuki 2010). In this method, a quartet of strains were examined at a time, and the corresponding phylogenetic tree was constructed for each gene segment. Then, the topologies of all quartet trees supported with a statistical significance were compared among segments. The reassortment events could be rec-ognized according to the pattern of topological difference among segments. Notably, although the reassortment events can be identified accurately, the computation com-plexity of the graph theory-based algorithm is tremendous, as the traversal of the phylogenetic tree with a part of strains will cost huge computing resources and time.
Obviously, the validity of the identified reassortment events with the phylogenetic tree-based methods is dependent on the reliability of the constructed trees. However, the false phylogenetic incongruence can be caused by the inaccurate construction of phylogenetic trees, such as inappropriate selection of evolution model (Keane et al. 2006), high level of homoplasy (Goloboff and Wilkinson 2018), long branch attraction (Li et al. 2007), insufficient sampling (Graybeal 1998), unreasonable data partition (Prosperi et al. 2011) and so on. To solve this problem, Svinti et al. developed two robust approaches to detect reassortments, namely MLreassort and Breassort, which can distinguish the reassortment-caused topological inconsistency from phylogenetic errors-caused topological inconsistency (Svinti et al. 2013). MLreassort is based on a maximum likelihood framework while Breassort is a Bayesian based approach. High precision and sensitivity were achieved when these two approaches detected reas-sortment events on both the small real data of influenza A sequences and the simulated data. However, the perfor-mance of these two approaches was not satisfactory when they analyzed the large datasets.
In conclusion, phylogenetic tree-dependent methods rely on the assumption that reassortants are distributed among the different clades of phylogenetic trees. These approaches are generally feasible to identify reassortment events across inter-subtypes of the influenza virus. They are accurate and sensitive to identify reassortment events even if the reassortant has a complicated evolutionary history. However, the reliability of phylogenetic tree con-struction is usually unsatisfactory when there is extremely incomplete data, and the low bootstrap probabilities and poor topology may lead to the obscure evidences for reassortment. Although some efforts were made to solve this problem, the feasibility of these methods is still limited by the computational cost from large scale data.
-
Ambiguous quantified benchmark for partitioning the phylogenetic clades and the extreme dependence of phy-logenetic reconstruction led more efforts on the identifi-cation of reassortment events without the phylogenetic trees.
The sequence distance between strains was commonly used in phylogenetic tree-independent methods. The Complete Composition Vector (CCV) was first employed to recognize reassortants by Wan et al. (Wan et al. 2007a, 2007b, 2008). In these algorithms, the calculated CCVs among different virus strains are core parameters, which are then used to assign diverse genotypes for related strains by different clustering methods. In their first algo-rithm (Wan et al. 2007a), the reassortment events can be identified by the genotypes which are assigned using the network modules coupled with the CCVs. As demonstrated in the study, this algorithm could infer the reassortment events with a large number of sequences accurately and rapidly. After that, the clustering method was improved by employing the minimum spanning tree (MST) and the Hierarchical Bayesian Modeling instead of the networks (Wan et al. 2008). As indicated in the evaluations, the CCV-based algorithms could successfully identify the reassortment events of the NP and PB2 genes for the H5N1 avian influenza virus. Another two algorithms were also developed with the sequence distance. Rabadan et al. constructed a statistical framework to estimate the likeli-hood of reassortments with the hamming distance in the third codon position for all sequences (Rabadan et al. 2008). The detected reassortment events of H3N2 strains with this algorithm were similar to the previous study. A reassortment identification algorithm was developed by Silva et al. based on the r-neighbourhood which are determined only by the genetic distances among sequences (de Silva et al. 2012). For each sequence, the set of r closest strains is defined as the r-neighbourhood for that sequence. 35 candidate reassortants of high quality were found by the algorithm with the large data sets of influenza virus whole genome nucleotide sequences. In addition, Chan et al. proposed that the pervasive reassortment in influenza virus can be detected with persistent homology (Chan et al. 2013).
Apart from the sequence distance, the other features were also employed to identify the reassortment events without the phylogenetic tree. Recently, a novel compu-tational algorithm HopPER (Yin et al. 2020) was proposed by Yin et al., which inferred the reassortment events by the random forest based on the prediction of the host tropism. 147 features generated from seven physicochemical prop-erties of amino acids (i.e. polarity, net charge, hydropho-bicity, normalized van der waals volume, solvent accessibility, polarizability and secondary structure) were used to infer the host tropism. For the full length and non-redundant amino acid sequences of different proteins, 280 out of 318 candidate reassortants were successfully iden-tified regardless of the completeness of the genomes. In addition, HopPER was more robust than the alternative reassortment identification algorithms (Karasin et al. 2000, 2002, 2006; Olsen et al. 2006; Khiabanian et al. 2009; Kingsford et al. 2009; Nagarajan and Kingsford 2011; de Silva et al. 2012).
-
In addition to the efforts which have working on the computational identification of influenza virus reassort-ments, several database tools were also developed to facilitate the computational related analysis for influenza virus reassortments.
Due to the diversity of influenza viruses that can reflect the possible reassortment events, the appropriate assigned genotypes are essential to identify and describe the reas-sortments of influenza viruses. FluGenome (http://www.flugenome.org/) was constructed by Lu et al., which enabled users to recognize the reassortment events by their developed genotype nomenclature (Lu et al. 2007). The available sequences for eight gene segments were retrieved from NCBI Influenza Virus Resource first; then the downloaded sequences were clustered into several lineages following criteria, in order to assign the strains into the nomenclatural genotypes. FluGenome provided three levels of information that included the segments (assigned lineage, strain name, segment, serotype, host, country, year, GenBank accession number, nucleotide sequence and sequence length), genomes (assigned genotype and acces-sion numbers of individual gene segments) and genotypes (all genotypes and the genomes assigned into each geno-type). With the analysis of more than 2000 complete viral genomes, 156 unique genotypes were revealed in Flu-Genome. Based on the developed genotypes, the reassort-ment events can be further detected by combining the epidemiologic information of the corresponding strains in the database. Unfortunately, FluGenome is no longer sup-ported, which is a grievous loss to the study of the influenza virus reassortments.
As the increasing number of studies have attempted to identify the reassortment computationally, a systematic, comprehensive online repository of reassortment events for influenza viruses is needed urgently. Our previous work developed FluReassort (https://www.jianglab.tech/FluReassort) (Ding et al. 2020), the first database that included all reported and published reassortment events. To facili-tate the investigation of the reassortment preference on the gene segment or the subtype of viruses, FluReassort also supported the reconstruction of reassortment networks for different subtypes of influenza viruses, which was based on the reassortment events retrieved from the extensive liter-ature. Total 3513 research papers published before July 2018 were retrieved from the PubMed database with a keyword combination of "subtype and (reassortment or reassortant or evolution or origin)", where "subtype" denotes the specific subtype of influenza virus such as H1N1. To provide the high quality reassortment events comprehensively, the reassortment events which were compiled manually from the given retrieved literature would be recruited in FluReassort only if they had both the phylogenetic analysis and clear reassortant and reassort-ment donor strains. As a result, 204 reassortment events were compiled based on 535 strains of 56 subtypes isolated from 37 different countries, which provides the metadata about the reassortant strain and reassortment donor strain, the inferred date, geographic region and host for reassort-ment, phylogenetic analysis methods and the PubMed IDs (PMIDs) of the corresponding references. FluReassort offered the most comprehensive information about reas-sortment events for influenza viruses in a structured way. The retrieval and exposition of the compiled reassortment events are implemented on the 'Home' page, while the 'Phylogenetic Analysis' and 'Reassortment Network' pages are designed to analyze the reassortment events. FluReassort has conducted a thorough compilation of the reassortment events for influenza viruses for the first time. The information provided by FluReassort can serve as a guide to future research, and facilitate data-driven explo-ration of the reassortments.
Bioinformatics Methods for Identifying Reassortments
Phylogenetic Tree-Based Methods
Phylogenetic Tree-Independent Methods
Database and Webtools for Influenza Virus Reassortments Identification
-
Both influenza virus genomic data and influenza virus-re-lated data have been exploded in the current big data era. Therefore, one of the challenges for researchers is how to process the influenza virus-related big data to improve the reliability of the influenza virus reassortment identification. Reasonable data preprocessing can bring several advan-tages to the computational reassortment identification. The volume of the data is decreased by removing the redundant information, which can reduce the computation time and the hardware requirements. For example, as indicated above, most phylogenetic tree-based methods are unavail-able for enormous amount of viruses. To address this sit-uation, a set of rational criteria, which eliminate the redundant strains sharing a close phylogenetic relationship, are needed urgently. An approach is to process the data by integrating the epidemiologic information and the pairwise sequence distance between strains. Non-redundant data also decreases the noise in the subsequent analysis with the identified reassortment events. For example, multiple influenza virus reassortants were generated in a reassort-ment event once; however, they were assigned with dif-ferent name or ID in a dataset. For the un-preprocessed data, this reassortment event might be regarded as different events in the computation process, which then influenced the subsequent analysis, such as the reassortment network construction. Lastly, the currently available influenza virus-related data are generated by high-throughput sequencing technologies, which have systematic sequenc-ing errors. Elimination of these errors will improve the accuracy of the reassortment event identification.
The integration of various types of influenza virus data is crucial to improve the reliability of the inferred reas-sortment events. As reviewed above, an effort was made by Yin et al. (2020), which identified the reassortments with features generated from seven physicochemical properties of amino acids. Although this algorithm has several limi-tations, an insight was provided in terms of the data-driven computational identification of influenza virus reassort-ments. Such as the previous studies have indicated that the protein structure can be influenced by the evolution of viruses (Nakajima et al. 2005). Thus, we infer that the combination between protein structure information and evolutionary profiles of influenza viruses can identify the reassortment events more sensitively and accurately. For example, similar to the Villa et al.'s work (Villa and Lassig 2017), an approach is to evaluate the mutations generated in the evolutionary pathways based on the function of different regions of viral proteins, which can be further employed to identify the reassortants from the phylogenetic trees. In summary, the reasonably processed data will guarantee accurate identification of the reassortment events.
-
As shown in Table 1, the update and even the download are no longer supported for most of the algorithms, which mainly results from the limited users of these algorithms. In our opinion, a reassortment identification algorithm should be developed aimed at the researchers from differ-ent fields. The usage for the researchers with non-computer background will be limited by the low practical and low validity of the algorithms. For example, the GiRaF algo-rithm not only needs to be compiled from the source code, but also requires to install a series of dependent libraries. Although the software developed by Dong et al. (2011) has a user-friendly interface, each identification process requires manual operation, which greatly reduces the effi-ciency of the software. Therefore, we suggest that the future reassortment identification algorithms encapsulate an easy-to-use pipeline and a user-friendly interface.
The other key difficulty to improve the identification methods is to define the rational thresholds to evaluate the heterogeneity of multiple gene segments between strains. The thresholds are hard to develop due to the complexity and diversity of the analyzed datasets. In addition, auto-matic estimation of cutoffs based on the analyzed data is also worth trying. For instance, the sequence distance cutoff that is used to recognize the heterogeneity of the intra-subtypes influenza viruses should be distinct from that for the inter-subtypes influenza viruses. Additionally, the development of the computational identification methods can focus on the self-adjusting estimation for the related thresholds. As reviewed above, most reassortment identi-fication methods are recursive, which leads the data structure to changing in each process. Therefore, the opti-mal adjustment for the cutoff is required for these algorithms.
-
Currently, there is lack of a benchmarking dataset to evaluate the performance of the developed reassortment identification algorithms. The mostly appropriate way is to generate a golden dataset that provides the reassortants, corresponding parental strains and the reassortment geno-mic segments confirmed by experimental methods in the laboratories. However, this work is impeded by the safety and ethical issues. An alternative computational way is to infer precise reassortment event that contain complete reassortment information by reliable data and appropriate methods. For example, the reasonable and effective clas-sification standard of the H5N1 viruses' HA segment can be used, which was proposed by World Health Organiza-tion (WHO), World Organization for Animal Health (OIE), and Food and Agriculture Organization (FAO) (WHO 2008, 2009, 2012; Smith and Donis 2015). In addition, several previous identified reassortment events are credible and include complete information, which are considered as the benchmarks in studies (Lam et al. 2013; Wu et al. 2013). In short, a benchmarking dataset is urgently needed for the development of the computational identification of the reassortments.
Optimal Processing of Big Data
The Practicality and Validity of the Reassortment Identification Methods
The Benchmarking Dataset
-
In this work, the computational identifications on influenza virus reassortment, which included the identification methods and the related database tools, were summarized comprehensively. In addition, the challenge and future prospects in computational identification of influenza virus reassortments were also illuminated. The reassortment identification methods were generally divided into two categories in terms of the dependence on phylogenetic tree. The phylogenetic tree-based methods recognize the reas-sortment events by investigating the structure incongru-ences among the phylogenetic trees of eight gene segments. Among these methods, manual comparison of the topolo-gies of phylogenetic trees coupled with epidemiologic information was the most commonly used. As the manual identifications were empirical and subjective, some auto-matic reassortment inferences based on phylogenetic trees were developed, which employed the graph theory, the statistics and so on. Although the reassortment events could be identified with these approaches accurately and sensi-tively, which is attributed to the use of the evolutionary history of related influenza viruses, the feasibility of these methods is tremendously limited by the reliability, time and computational cost of the phylogenetic tree construc-tion. In this case, several efforts were made to recognize the reassortants without phylogenetic trees. The phyloge-netic tree-independent methods detected the reassortment events primarily by using the significant differences of strain distances among multiple gene segments, which can be implemented on large amount virus strains with low computational complexity. These distances were based on both the genomic sequences and the physicochemical properties of amino acids. However, the quality of nucleotide and amino acid sequences could greatly influ-ence the identification performance. For the reviewed reassortment identification algorithms, the performance were regrettably not compared because the most algorithms are unavailable and the benchmark dataset is lacking. Therefore, we only summarized the actual use experience of the four obtained software in Table 1. Based on the principles of the algorithms, we also give some suggestions on different application scenarios of these algorithms for both bioinformatic and virological researchers. Firstly, the software developed by Dong et al. is the only algorithm that has a friendly interface, which is more suitable for researchers with little bioinformatics background (Dong et al. 2011). In general, the phylogenetic tree-independent methods are more efficient to identify the reassortments from the large-scale data compared to the phylogenetic tree-based methods which are quite time-consuming when constructing the phylogenetic tree. However, for a small number of genomic sequences (usually about 100 sequen-ces), the phylogenetic tree-based methods infer the reas-sortments more accurately. In addition, the methods proposed by Nagarajan and Kingsford(2008, 2011), Wan et al. (Wan et al. 2007a, 2007b, 2008), Rabadan et al. (2008) and de Silva et al. (2012) can identify the reas-sortment events based on partial genomic segment sequences of influenza viruses. The amino acid sequences of genomic segments can be processed by either FluResort or HoPER, and HoPER is more suitable for inferring the reassortments in the same host.
On the other side, as an increasing number of studies on identifying influenza virus reassortments, two databases i.e. FluGenome and FluReassort were developed, providing valuable information related to the influenza virus reas-sortments. In summary, a universal and valid computa-tional method for reassortments identification doesn't exist recently. The most appropriate scheme can be designed, which depending on all information of the analyzed data, such as the amount of strains, the diversity of strains and so on. We hope this review can serve as a guide to reasonably identify the reassortments for diverse influenza virus datasets.
-
This work was supported by the National Natural Science Foundation of China (31801101 to X.D., 31671371, 32070678 to T.J.); the CAMS Initiative for Innovative Medicine (CAMS-I2M, 2016-I2M-1-005, 2020-I2M-2-003 to T.J.).
-
Conflict of interest The authors declare that no competing interests exist.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: