














-
Dear Editor,
Coronavirus Disease 2019 (COVID-19) outbreak caused by sever acute respiratory syndrome coronavirus 2 (SARS-CoV-2) presents a global pandemic which has resulted in more than 4 million people death in the world. In China, the initial transmission of COVID-19 has already been blocked by strict control strategies and effective treatment for patients. All local COVID-19 outbreaks after April 2020 were related to overseas importing. Up to February 2021, 207 imported COVID-19 cases have been reported in Tianjin, China (Tianjin Health Commission, 2021).
SARS-CoV-2, a kind of positive-sense single stranded RNA virus and belongs to the Betacoronavirus genus of the Coronaviridae family. Fourteen open reading frames (ORFs) constitute the majority of the ~ 2.9-kb SARS-CoV-2 genome, which encodes four structural proteins including nucleocapsid protein (N), membrane protein (M), spike protein (S) and RNA independent RNA polymerase (RdRp) and other non-structural proteins (Lu et al. 2020). The intrinsically high error rates of RdRp result in the stochastic introduction of mutations during viral genome replication (Liu et al. 2021). SARS-CoV-2 has evolved into two main lineages according to genome-based typing method established by China CDC (China CDC Lineage) and PANGO Lineages method (PANGO Lineage) (Rambaut et al. 2020; Wu et al. 2020; Yang and Xu 2021). The S/A-lineage defined by two nucleotides (8782T, 28144C) mainly circulates in Asia and the L/B-lineage defined by 8782C and 28144T mainly circulates in Europe and America. B.1.1.7-lineage/501Y.V1 mutation exhibited a rapid increase in its range and incidence, and other variants emerged in succession namely B.1.351-lineage/501Y.V2 from South Africa, P.1-lineage/501Y.V3 from Brazil, and B.1.207-lineage from Nigeria (Hodcroft et al. 2021; Oude Munnink et al. 2021). In this study, high through-put whole genome sequencing (WGS) was used to sequence all the imported SARS-CoV-2 samples for showing the genome characteristics and helping to design control strategies.
The study involved 94 throat swabs and one swab collected on the outer packaging of cold-chain food products (sample ID: TJ_20TF_FH158) from March 2020 to February 2021 (Supplementary Table S1). The nucleic acid was extracted using viral RNA extraction kits and instruments (Xi'an Tianlong Science and Technology Co., China). WGS was carried out using Nextera XT DNA Library Preparation Kit and MiniSeq System (Illumina, USA). Fifty-six SARS-CoV-2 genomes with the coverage higher than 98% and the sequencing depth higher than 100× were obtained by de novo assembly.
For nucleotide variation analysis, 309 single nucleotide polymorphisms (SNPs) and 14 deletion/insertion sites were found in all 56 SARS-CoV-2 genomes by using Bowtie2 software (Supplementary Figure S1). Less than ten SNPs were identified from each of genomes collected from March 2020 to June, and more than 17 SNPs were found from genomes collected from October 2020 to February 2021 (Supplementary Table S1). According to China CDC Lineages and PANGO Lineages, T8782C and C28144T were two specific SNPs of S/A-lineage. In this study, four sequences fell into the S/A-lineage, and 34 SNPs and 3 deletion/insertion sites were found in these strains. The other 52 sequences all fell into the L/B-lineage. Four SNPs (C241T, C3037T, C14408T and A23403G) were detected as the characteristics of L-lineage European branch (China CDC lineage method)/B.1-lineage (PANGO Lineage) in 50 genomes. Nine sequences had three SNPs (G28881A, G28882A and G28883C) which were detected as the characteristics of L-lineage European branch I/B.1.1-lineage. The other nine sequences had two SNPs (G25563T and C1059T) which were characteristics of L-lineage European branch II.1. Four sequences had the three SNPs (G25563T and C2416T) which were detected as the characteristics of L-lineage European branch II.2. Five sequences had the two SNPs (G25563T and C18877T) which were detected as the characteristics of L-lineage European branch II.3.
For phylogenetic analysis, 1494 high-quality and high-coverage SARS-CoV-2 genomic sequences were downloaded from Global Initiative on Sharing Avian Influenza Data (GISAID) and aligned by MAFFT v7.42 and IQ-tree v2.1.2 (Shu and McCauley 2017). 895 sequences of S/A-lineage included 18 strains which all had the N501Y and L452R mutation of S protein and can constitute an individual sub-lineage, named as the A_501Y lineage including TJ_A371, TJ_21TF_FH07 and TJ_A106 (Fig. 1A). Two genomes (EPI_ISL_801441, EPI_ISL_801442) were closely related to TJ_A371, TJ_21TF_FH07 and TJ_A106. The original region of these strains included Belgium, Turkey, Mayotte, Spain, et al, and the date of specimen collection ranged from December 2020 to January 2021. The other 599 sequences of L/B-lineage included the B.1.1.7/501Y.V1 variant (sample ID: TJ_B449_2) which was the first detection of a major international variant in Tianjin Municipality in January 2021 and was closely related to two genomes, EPI_ISL_1060597, EPI_ISL_1018092, collected from France and Ghana, respectively. Four genomes (EPI_ISL_806290, EPI_ISL_428910, EPI_ISL_1383183, EPI_ISL_1111200) were closely related to TJ_20TF_FH158 according to phylogenetic analysis, all fell into the B.1.1.1-lineage and shared four specific SNPs, C4002T, G10097A, C13536T and C23731T, and other five unique SNPs (C1612T, G3606T, C7772T, C25665T and C26600T) were found in genome of TJ_20TF_FH158.
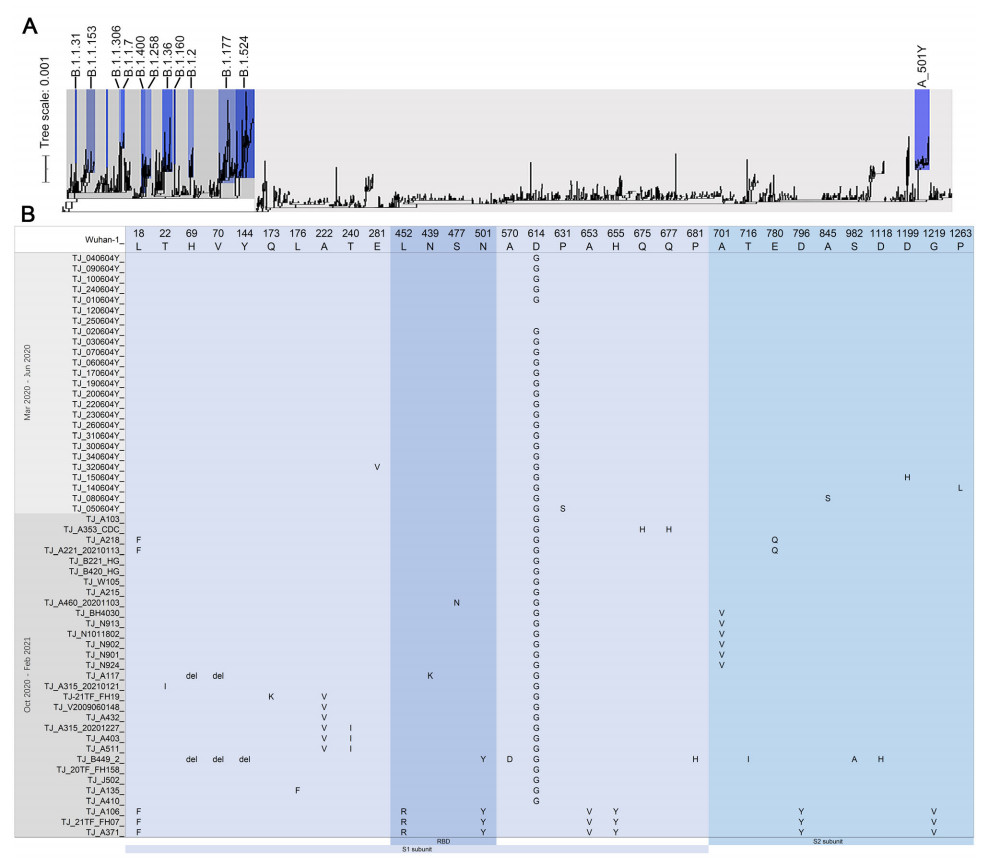
Figure 1. A Phylogenetic analysis of the SARS-CoV-2 isolated in Tianjin, China. Maximum likelihood phylogeny inferred using 56 genome sequences of SARS-CoV-2 generated in this study and 1494 sequences already deposited on the GISAID database. The tree is rooted between A-lineage and B-lineage. B Variant sites of S protein amino acid identified in 56 SARS-CoV-2 genomes obtained in this study.
S protein comprises S1 subunit locating at the N terminal and S2 subunit locating at the C terminal. The receptor-binding domain (RBD, S protein aa319-541) of S1 subunit binds to the human angiotensin I converting enzyme 2 (ACE2) receptor which mainly depends on the receptor binding motiff (RBM, S protein aa438-506) region, and S2 subunit helps virus fusion into cells (Rathnasinghe et al. 2021). RBD is also a target of the SARS-CoV-2 neutralizing antibody, and mutations in this region may affect the neutralizing titer of antibody (Massacci et al. 2020). 29 amino acid (aa) variants and three aa deletion sites of spike protein were found in all 56 genomes, and 22 variants of them located in the S1 subunit including four variants in the RBD region of S1 subunit (Fig. 1B). Seven aa variants including L18F, L452R, N501Y, A653V, H655Y, D796Y and G1219V were identified in TJ_A371, TJ_21TF_FH07 and TJ_A106 of A_501Y lineage (Fig. 1B). For B.1.1.7/501Y.V1 variant, 28 nucleotide variants as the characteristics were identified in the genome of TJ_B449_2 including ten aa variants/deletion of S gene (Fig. 1B), and other six unique SNPs including C2110T, G2914T, T7984C, G10887A, C14120T, C19390T and nucleotide deletion of 27792–27794 and 28271 were found in this genome. Nucleotide deletions of 26160–26167, 27386 and 28248–28253 were found in genomes of TJ_21TF_FH07 and TJ_A371 (Supplementary Table S2).
Initial low percentage of sequences in S/A-lineage exhibited a slight increase from December 2020 to the beginning of 2021 and were discovered in many countries worldwide with several aa mutations of S protein. In this study, N501Y and L452R mutants locating in RBD region of S protein were found in three sequences belonging to A_501Y lineage. L452R may decrease the sensitivity of the virus to neutralizing antibody. N501Y mutation may affect the immunogenicity of the virus (Liu et al. 2021; Gu et al. 2020). N501Y variant of A lineage have been identified in Turkey, the United Kingdom, France, Denmark, Niger, et al (Li et al. 2020). The pathogenicity and transmissibility of A_501Y mutation were still unknown.
Two waves of COVID-19 outbreaks emerged in Tianjin Municipality. At the beginning of 2020, the outbreak was mainly resulted by small-scale local area transmission. In November 2020, the outbreak was associated with imported cold chain products emerged in local communities. Numbers of accumulated COVID-19 positive travelers from overseas in Tianjin continually rise, and the risk of imported COVID-19 outbreaks remains. Sustained SARS-CoV-2 genome sequencing and monitoring of variants can be used to enrich genome data of SARS-CoV-2 which is of great significance for implementing prevention and control strategy of COVID-19 outbreak and tracing the infection sources (Chen et al. 2021; Eden et al. 2020).
HTML
-
This work was supported by Tianjin Municipal Commission of Health COVID-19 Prevention and Control Technology General Project (No. 2020xkm01 and No. 2020xkz01), and Science and Technology Program of Tianjin, China (No. 20JCZDJC00130).
-
The authors declare that they have no conflict of interest.
-
Additional informed consent was obtained from all patients for whom identifying information is included in this article.

 DownLoad:
DownLoad: