












-
Dear Editor,
Clinical outcomes of SARS-CoV-2 infection are highly heterogeneous, ranging from symptoms-free infection to death of the patient (Park et al. 2020). Although the outcome can be explained to some extent by age and comorbidities, genetic factors have also been linked to the prognosis of SARS-CoV-2 infection (Hu et al. 2021; Liu et al. 2020; Maya et al. 2020; Pairo-Castineira et al. 2021; The Severe COVID-19 GWAS Group 2020; Zeberg and Pääbo 2020). To this date, the association of nine single nucleotide polymorphisms (SNPs) at genome-wide significance level has been reported in the Genome Wide Association Study (GWAS) catalog for severe COVID-19 outcome phenotype (MONDO_0100096) (GWAS catalog 2021). These SNPs were identified in two studies involving 4378 COVID-19 patients of European, 176 admixed American, and 62 of South Asian ancestry (Pairo-Castineira et al. 2021; The Severe COVID-19 GWAS Group 2020). A leading association across these studies is found in the 3p21.31 region, specifically near genes LZTFL1 and SLC6A20 (Pairo-Castineira et al. 2021; The Severe COVID-19 GWAS Group 2020), and rs11385942 variant from this region also shows the lowest heterogenous P value in a worldwide cohort of 49 studies from 19 countries (The COVID-19 Host Genetics Initiative and Ganna 2021).
While the primary attention in previous studies was to identify genetic associations with the severity of the COVID-19 disease, we decided to evaluate the already reported genome-wide significant associations regardless of the disease severity in a representative sample of 2692 individuals from the population of Latvia. Additionally, we aimed to further examine the corresponding loci to identify new associated polymorphisms in a region with positive signals. The study included all 475 COVID-19 patients recruited to the Genome Database of Latvian Population (LGDB) (Rovite et al. 2018) from June 2020 to January 2021. This cohort consisted of 146 hospitalized patients, while the rest represented less severe symptoms or asymptomatic cases. In total, 2217 controls were selected from LGDB, representing the group of the general population and disease-specific biobank participants from the Latvian population with available genome-wide genotyping data. This study was approved by the Central Medical Ethics Committee of Latvia (No. 01-29.1.2/928). The mean age of cases and controls were 47.3 [standard deviation (SD) = 17.8] and 54.9 (SD = 13.1) years old, respectively. The proportion of females was 64.5% in the case group and 62.0% in the controls. After the genotype quality control and imputation using the TOPMed r2 imputation server, we performed an association study with sex and age as covariates. Association was performed with PLINK 1.9 (Chang et al. 2015) according to the parameters defined in SAIGE software (Zhou et al. 2020), adding the first 20 principal components (PCs) to control for population stratification.
We selected a total of nine significantly associated SNPs reported in the GWAS catalog from two previously published studies analyzing the association between severe COVID-19 cases and population controls (Pairo-Castineira et al. 2021; The Severe COVID-19 GWAS Group 2020). Table 1 summarizes the list of the selected SNPs. No significant deviation in allele frequencies (AF) was found between the control group and the global AFs from the 1000G Phase 3 reference set (The 1000 Genomes Project Consortium et al. 2015). Out of the nine selected variants, we identified three significantly associated LZTFL1 gene polymorphisms rs71325088 [Bonferroni adjusted P-value (Padj) = 0.007, odds ratio (OR) = 1.46 95% confidence interval (95% CI) 1.17-1.81], rs11385942 (Padj = 0.005, OR = 1.47 95% CI 1.18–1.820) and rs73064425 (Padj = 0.007, OR = 1.45 95% CI 1.17–1.80) after applying the Bonferroni correction for multiple testing (Table 1). We observed a high degree of linkage disequilibrium (LD) between all three SNPs reflected by almost identical frequencies in case and control groups. These variants were also the most significant (Phet = 7.2 × 10−25) in the worldwide meta-analysis of individuals with the SARS-CoV-2 infection, hospitalization, and critical illness (The COVID-19 Host Genetics Initiative and Ganna 2021). All of these variants are believed to represent the region to be a remnant of Neanderthal gene pool introgression into the modern human population (Zeberg and Pääbo 2020). LZFTL1 gene has been implicated in ciliogenesis and intracellular trafficking of ciliary proteins, probably impacting airway epithelial cell function (Promchan and Natarajan 2020; Shelton et al. 2021). Leading polymorphism rs11385942 is located at 618 bp upstream and 670 bp downstream from LZTFL exons as well as 1800 bp upstream and downstream from CTCF and transcription factor binding sites (GTEx Consortium 2018).
Chr SNP ID Risk allele Mapped genes COVID-19 patients (n = 475) vs. controls (n = 2217) Hospitalized COVID-19 patients (n = 146) vs. controls (n = 2217) P/Padj AF (case/ctrl) OR (95% CI) P/Padj AF (case/ctrl) OR(95% CI) GWAS catalog OR (95% CI) 3 rs71325088 C SLC6A20, LZTFL1 0.0007/0.007 0.13/0.09 1.46 (1.17–1.81) 0.00001/0.0001 0.16/0.09 2.10 (1.50–2.92) 1.9a (1.73–2.0) 3 rs11385942 A LZTFL1 0.0005/0.005 0.13/0.09 1.47 (1.18–1.82) 0.000006/0.00006 0.16/0.09 2.14 (1.54–2.99) 2.11b (1.70–2.61) 3 rs73064425 T AC099782.3, LZTFL1 0.0008/0.007 0.13/0.09 1.45 (1.17–1.80) 0.00001/0.00009 0.16/0.09 2.13 (1.52–2.97) 1.7a NR 6 rs143334143 A CCHCR1 0.7/1 0.09/0.09 0.94 (0.73–1.22) 0.4/1 0.10/0.09 1.18 (0.78–1.80) 1.3a (1.27–1.48) 9 rs657152 A ABO 0.1/0.891 0.47/0.44 1.13 (0.98–1.31) 0.1/1 0.48/0.44 1.22 (0.95–1.55) 1.32b (1.20–1.47) 12 rs6489867 C OAS1, AC004551.1 0.9/1 0.30/0.31 0.99 (0.85–1.16) 0.7/1 0.29/0.31 0.96 (0.73–1.25) 1.2a (1.14–1.25) 19 rs2109069 A DPP9 0.01/0.115 0.30/0.27 1.22 (1.04–1.44) 0.1/1 0.30/0.27 1.22 (0.94–1.59) 1.2a (1.19–1.31) 19 rs11085727 T TYK2 0.03/0.235 0.31/0.28 1.20 (1.02–1.40) 0.1/1 0.32/0.28 1.23 (0.95–1.60) 1.2a (1.18–1.31) 21 rs13050728 T AP000295.1, IFNAR2 0.6/1 0.39/0.38 1.04 (0.90–1.21) 0.06/1 0.43/0.38 1.28 (0.99–1.64) 1.2a (1.16–1.28) Bold represents the significant P-values after Bonferroni correction
aFromPairo-Castineira et al. 2021
bFrom The Severe COVID-19 GWAS Group 2020.
Bold represent the significant P-values. Chr, Chromosome number; SNP, Single nucleotide polymorphism; AF, Allele frequency; Padj, Bonferroni adjusted P-value; ctrl, controls; OR, Odds ratio; CI, Confidence interval; NR, not recordedTable 1. Association of the selected single nucleotide polymorphisms and SARS-CoV-2 infection
Even though the primary analysis was performed on the case group that included all patients regardless of severity, there was an obvious bias toward the inclusion of symptomatic patients as SARS-CoV-2 was more likely to be tested in these patients than asymptomatic cases. We also tested the association of the selected SNPs with disease severity in our study group using hospitalized patients as cases against the same group of controls (Table 1). All three SNPs from the 3p21.31 region displayed a stronger association in the frame of this comparison (Padj = 0.00006, OR = 2.14 95% CI 1.54–2.99) and 4.2 times higher homozygote prevalence for rs11385942, supporting the notion that this locus increased the risk for respiratory symptoms. Studies that account for participants' exposure to the infection are needed to answer this question. However, the other SNPs included in the study did not reach statistical significance. The OR for almost all SNPs (excluding rs6489867) was similar to those published previously (Pairo-Castineira et al. 2021; The Severe COVID-19 GWAS Group 2020) (Table 1). Probably larger sample size is needed to replicate other loci. It should be noted that the 3p.21.31 locus is the only replicated locus out of all 28 COVID-19 variants released in GWAS catalog to date, representing variants above P = 5 × 10−8 level.
We also explored the 500 kb regions around the rs11385942 to evaluate the association of other SNPs with the main trait in our study. In total nine 3p21.31 locus polymorphisms, rs2191031, rs3774641, rs72893671, rs35896106, rs13071258, rs34668658, rs17763742, rs17763537 and rs73062389 displayed lower P-value in our cohort compared to the leading rs11385942, with the strongest association estimated for rs2191031 (P = 0.00005, OR = 1.40 95% CI 1.19–1.64) located in the LZTFL1 gene (Fig. 1). Among these newly discovered variants, rs2191031, rs3774641 are located in a promoter flank and rs73062389 in CTCF region, while rs35896106 overlaps enhancer of SLC6A20 gene. The prevalence of rs2191031 is 2.3 times higher in our population compared to replicated polymorphisms. To date, no function or clinical relevance has been ascribed for this variant; however, rs2191031 is equivalently associated with differential expression of the LZTFL1 gene in the esophagus mucosa (P = 10−5, normalized effect size = − 0.22) (GTEx Consortium 2018) and multiple other tissues pointing to similar etiology as replicated variants (Shelton et al. 2021). However, it is important to note that none of these nine variants have a lower P-value than rs11385942 in a cohort of hospitalized COVID-19 patients.
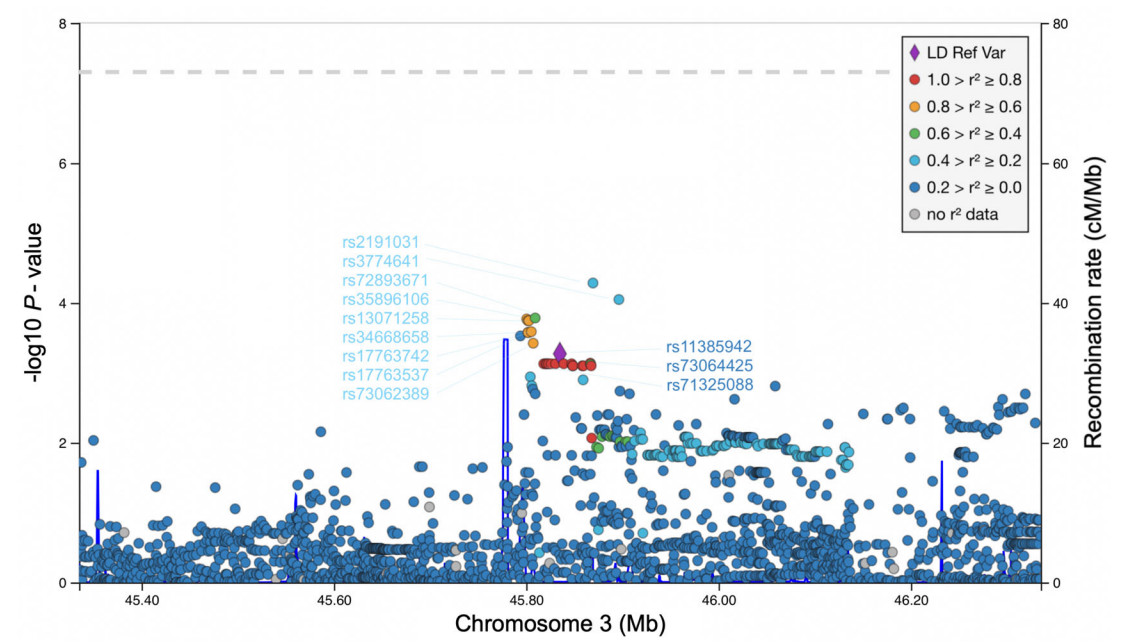
Figure 1. Manhattan plot of 1 Mb region at 3p21.31 locus. rs11385942 variant is emphasized with a diamond shape and set as a reference for LD estimation with other SNPs in the region. Rs codes for three SNPs selected for replication are depicted in dark blue, while rs codes for other SNPs displaying lower P-value in our association study are in light blue. The dotted line represents the common genome-wide significance threshold. LD, linkage disequilibrium; SNP, single nucleotide polymorphism.
Using the retrospectively collected samples from a population-based biobank has some limitations. We cannot assess the exposure to SARS-CoV-2 for this group of people and match that with the case group, as we do not have the information about the possible rate of infection among this group. However, such a design does not increase type I error, and this study did not aim to find genetic factors facilitating protection against SARS-CoV-2 infection, in which such a design would not be appropriate. Another shortcoming was that most patients included in this study were recruited in December 2020, and extensive phenotype data, including follow-ups, were not yet available for analysis. Such data would allow to elaborate the interaction between different clinical data and genotype and include the development of post-COVID-19 complications as an essential phenotype for association study.
In conclusion, we demonstrate supportive evidence for the involvement of human 3p21.31 locus in the pathophysiology of COVID-19 disease using an independent cohort of patients and controls from the Latvian population. It highlights the importance of this genomic region for genetic risk estimation in relation to SARS-CoV-2 infection and the robustness of proper genetic association studies for replication purposes. Notably, the results presented here provide a preliminary indication of variants with possible functional effects and calls for further studies exploring the validation of these variants.
HTML
-
This study was funded by the Ministry of Education and Science, Republic of Latvia, project "Establishment of COVID-19 related biobank and integrated platform for research data in Latvia", project No. VPP-COVID-2020/1-0016. We acknowledge The Boris and Inara Teterev Foundation for support to Riga Stradins University inpatient sample collection. The authors acknowledge the Latvian Biomedical Research and Study Centre and the Genome Database of the Latvian Population for providing the infrastructure, biological material, and data.
-
The authors declare that they have no conflict of interest.
-
This study was approved by the Central Medical Ethics Committee of Latvia (No. 01–29.1.2/928).

 DownLoad:
DownLoad: