











-
Enterovirus 71 (EV71) belongs to the genus Enterovirus, family Picornaviridae. Since the first report of this virus (24), outbreaks of EV71 infection have occurred throughout the world (2, 9), especially in the Asia-Pacific region (6, 9, 19, 25). Most manifestations are mild childhood exanthema known as hand-foot-and-mouth disease (HFMD) (6, 9, 19, 25). However, in a few cases, EV71 may also cause severe neurological diseases and even the deaths of infected children, and can resulted in panic in the general population (5, 7).
Positive selection is defined as a significant excess of nonsynonymous (dN) over synonymous (dS) nuc-leotide substitutions (3). Specifically, when negative selection acts on a pool of mutants this might remove the mutations resulting in amino acids changes. In contrast, under positive selection, variants were per-mitted so that they could replace the original population. In other words, positive selection might favor their survival. For example, the haemagglutinin (HA) gene is one major surface glycoprotein of influenza A viruses. Previous studies have proved that positive selection pressures acting on HAs of H1, H3 and H5 subtypes were responsible for the evolution of the antigenic sites and receptor-combining sites (4, 10, 13).
VP1 is the most exposed and immunodominant of the picornavirus capsid proteins (23). Based on the estimated phylogenetic relationships of the VP1 proteins, three genotypes and eight subgenotypes have been identified (2, 6, 17, 19). Most neutralization sites that have been identified on the picornaviruses surface reside in the VP1 protein (18, 20). In addition, it is more variable than other regions in the viral genome (1). Therefore, positive selection pressure on VP1 proteins might affect the variability of the antigenic sites and modulate the virus-receptor interaction.
In this study, we present a study regarding the positive pressures acting on VP1s of EV71 viruses. Our analysis provides evidence of the evolution of key amino acids sites in VP1s that might correlate to biologically important regions of EV71 viruses. It would help to design and develop vaccines.
HTML
-
Sequence data were downloaded from GenBank directly, which were updated in April 28th, 2008. After removing some short sequences, we included a total of 633 VP1 sequences into this analysis. These sequences were further divided into several smaller datasets according to isolation location and time (Table 1).

Table 1. Datasets into this analysis
Multiple sequence alignment was performed manually for each dataset. A Maximum Likelihood (ML) tree was built for each data subset using PHYML v2.4.4 for Windows (11). Ts/tv ratio and proportion of invariable sites were both estimated rather than treated as fixed.
Site-by-site positive selection analysis was per-formed for each data subset using the Fixed Effect Likelihood (FEL) method in the HyPhy software Package (15). ML trees obtained in the previous step were used in the analysis. An HKY85 model was selected as the best model of sequence evolution for the data. A global dN/dS ratio was estimated by a codon model obtained by crossing MG94 and HKY85. A two-rate FEL model was applied allowing dN and dS to be adjusted across sites. P-values < 0.1 were considered to be significant.
At last, we constructed the 3-D structures of VP1 proteins of typical EV71 virus, Coxsackievirus B4 (CVB4) and Human rhinovirus 1A (HRV1A) through the online service provided by ESyPred3D (16). By structural comparison, we tried to locate the positions of the positively selected sites identified in the previous step onto the 3-D models and to identify their possible biological functions.
-
Our results showed that four sites in the VP1 regions of EV71 viruses were detected to be under positive selection pressure (Table 2). Among the four positively selected sites, position 145 was found to be positively selected for viruses from Malaysia, Singapore, Taiwan and USA, whereas the other three positions were only under positive selection for viruses from a certain region (Table 2).

Table 2. Positively selected sites and their amino acids polymorphism
Amino acid polymorphism analysis revealed that at position 58, most of the viruses from Australia isolated since 1999, Japan 2000-2003, Malaysia 1997 and 2000-2003, Taiwan 1999-2003 and USA before 1990 had T, while other viruses had A instead (Table 2). E was observed to be dominant at position 98 for many viruses, except for those from Australia, Malaysia 1997-1999 and USA before 1980, some of which had K (Table 2). Viruses with E, G and Q at position 145 accounted for 99.53% of the total. Among them, 396 viruses (62.76%) had E at this position (Table 2). Except for viruses from the USA collected between 1980 and 1989, most viruses had S at position 241, where M and L were sporadically detected (Table 2).
More importantly, the results of structural com-parison between EV71 and CVB4 showed position 98 was located in the BC loop (Fig. 1). Besides, com-paring the 3-D structures of VP1 proteins of EV71 and HRV1A, we found both positions 145 and 241 were located in the canyon rim region (Fig. 2).
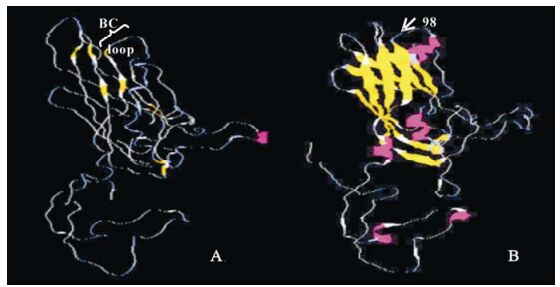
Figure 1. Structural comparison of CVB4 and EV71. A was the predicted 3-D structure of the VP1 protein of CVB4, and B was the predicted 3-D structure of the VP1 protein of EV71. All structures were displayed in Rasmol 2.7.1.

Figure 2. Structural comparison of HRV1A and EV71. A:the predicted 3-D structure of the VP1 protein of HRV1A; B:The predicted 3-D structure of the VP1 protein of EV71. All structures were displayed in Rasmol 2.7.1.
-
EV71 virus is one of the major etiological agents of HFMD that may cause serious neurological compli-cations. In previous studies, a number of genotypes and subgenotypes have been identified based on the phylogenies of VP1s (2, 6, 17, 19). In addition, recom-bination events among different EV71 and coxsac-kievirus A16 (CA16) viruses have been also found and were reported to play important roles in the emergence of the various EV71 subgenotypes and the altered pathogenic potentials (8, 27).
Here we performed a selection pressure analysis of the VP1 proteins. Our results showed that although most sites of VP1 proteins were negatively selected or under neutral evolution, four positions were under positive selection pressure. Position 58 was one of the six sites that were previously reported to display co-variability patterns and the co-variance of these positions was correlated to the subgenotypes (9). Through structural comparisons, position 98 was located in the BC loop (Fig. 1), which was one of the five VP1 loops forming an important neutralizing antigenic site (21). In addition, BC loop might also be involved in modulating the interaction between the virus and the host (22). Positive selection pressure on this position might imply EV71 viruses were under immunological pressure from the hosts and the possibility of potential variability at this position. In addition, positive selection pressure acting on position 98 might correlate to the enhanced attachment ability of the EV71 viruses to their hosts. Position 228 of many human rhinoviruses was located at the rim of the canyon and was believed to be one of the five amino acids determining receptor characteristics (14). Positions 241 of EV71 was located at a position similar to position 228 of many human rhinoviruses and position 145 was also identified to locate in the canyon rim. Therefore we conjectured that they might be involved in deciding in the receptor specificity of the EV71 viruses.
The A170V mutation was reported to be associated with the increased neurovirulence of these viruses and appeared to be a marker for a neurovirulent lineage(19). However, our results showed that this position was not under positive selection. Furthermore, only a few isolates from Australia since 1999 had this mutation, while all other viruses did not exhibit this substitution. Instead, position 170 was negatively selected for al-most all the datasets except for one consisting of Australian isolates. This might imply mutation at this position would be harmful and had been purified.
However, our results showed that many positions in the VP1 regions were highly conserved. Even at the four positively selected positions, viruses from different regions and time displayed little amino acid polymorphism (Table 2). Nonetheless, because of the presence of positive selection pressure, the possibility of potential amino acid variability still existed. There-fore, it is necessary to keep wide surveillance on the molecular epidemiology and genetic variability of EV71.
As viruses consisted of RNA genomes, only a few sites of VP1s of EV71 received positive selection pressures. This was not similar to the case observed in influenza viruses of H5 and H3 subtypes where several sites in the antigenic sites were under positive selection (4, 13). We thought this might be caused by two reasons. On one hand, there has been no vaccine available to prevent the EV71 infection to date. On the other hand, EV71 viruses mostly infect humans so far and they do not need to overcome the host barriers presented to the influenza viruses.
EV71 viruses have been circulating in some pro-vinces in southern China from February, 2008. Up to May 9th, 2008 they had caused 34 fatalities of children (12). However, our analysis showed that the EV71 viruses isolated from China had not been under positive selection from 1987 to 2007 and they displayed little difference in amino acid polymorphism at the positively selected positions in their VP1 genes com-pared to those of viruses from other regions (Table 2).
In conclusion, although four positions of VP1s proteins of EV71 viruses were under positive selection, they displayed limited amino acid polymorphism at these sites. Additionally, Chinese isolates did not show much variance in the VP1 region compared to those of viruses from other regions. However, positive selection pressures working on the VP1s and the potential recombination events make it possible for the EV71 viruses to undertake further variance that leads them to gain genetic advantages that favor their survival. Therefore, it is necessary to keep track of the molecular epidemiology and evolution of the EV71 viruses.


 DownLoad:
DownLoad: