











-
Reoviruses are distributed widely in aquatic environments and have been isolated from a wide range of aquatic organisms. Grass carp reovirus (GCRV) is currently one of the most serious pathogens threatening grass carp Ctenopharyngon idellus production with high mortality in China[29]. The virions consist of a double-layered protein capsid containing 11 dsRNA genomic fragments[38]. GCRV was assigned to the genus Aquareovirus of the family Reoviridae by the international committee on Taxonomy of Viruses (ICTV) in 1991[10]. It differed from orthoreovirus in a number of characteristics such as absence of an antigenic relationship and unequal numbers of genomic segments[2]. Grass carp kidney cells (CIK) are sensitive to GCRV infection and have been adapted for viral amplification and so were employed as an ideal in vitro model in the study of grass carp hemorrhagic disease[26, 35] caused by GCRV infection.
Among the defense systems of organisms, RNA interference (RNAi) is a new-found ancient mechanism of gene regulation with important antiviral roles[26]. A core feature of RNA silencing detected in all organisms is the production of 21-to 26-nt small RNAs cleaved by the endoribonuclease Dicer[13] and the role of RNAi as an important antiviral approach has been reported in most multicellular organisms[36]. Fruit fly Drosophila melanogaster is a model system for studying innate immunity, including antiviral host defense[11]. Long dsRNA, emerging during viral infection and replication or after transposition of mobile genetic elements, can efficiently cause gene-silencing in Drosophila through processing to small interfering RNAs (siRNAs) by the trigger nuclease Dicer[24, 33]. siRNAs targeting specific homologous mRNA molecules cause these mRNA to be degraded by ribonucleases[15]. Dicer is the heart of the RNAi molecular machinery and is responsible for gene silencing and micro-RNA (miRNA) processing for gene regulation[9, 27]. Similar to Drosophilar, plants also use Dicer-like proteins as the central enzymes of RNA silencing, which regulate gene expression and mediate defense against viruses[5]. Null alleles of Dicer result in embryo death in fish and mice[12], which signifies the role of Dicer in cellular basic functions besides RNAi. In all these multicellular organisms, RNAi can be efficiently triggered by either long dsRNAs or 21 to 23-nt-long siRNAs[3]. RNAi mechanism has been increasingly explored in invertebrates and mammalian cells for specific gene silencing[22].
Although GCRV can efficiently infect and replicate in CIK cells, recent studies found that chemically-synthesized siRNA targeting the sequence of its dsRNA genome could efficiently inhibit viral replication in CIK cells[23]. Transgenic Gobiocypris rarus bearing specific siRNA toward the viral genome was resistant to GCRV challenge[21]. These results implied that initiation of GCRV infection could be blocked by the effector nuclease of RNAi pathway downstream of initiator endonuclease Dicer. Similar conclusions were drawn from analysis of mammalian reovirus and avian reovirus[17-19]. These studies supported the premise that a siRNA-mediated RNAi pathway could functionally block the infection and replication of reovirus in CIK cells. We thus deduced that there shouldn't be any virus-specific siRNA in cells during efficient reovirus replication. dsRNAs generated from some RNA viruses were shown to be resistant to Dicer digestion due to their specific secondary structure[7, 32], while more viruses survived from RNAi by encoding RNAi suppressors[8, 25]. To define which strategy has been adopted by GCRV to escape the RNAi-mediated defenses, a Northern blot analysis was designed in this study to monitor the GCRV-specific small molecular RNAs in infected CIK cells as well as cells transfected with extracted genomic dsRNA of GCRV.
Although it was believed that input reovirus dsRNA genome might remain within its inner capsid throughout the viral life cycle to evade cellular innate immune response, it could be that minute amounts of accidentally uncoated or packaged genome, as well as secondary structure on mRNA, could be exposed to the cellular dsRNA-dependent enzymes[16]. Using a mouse specific monoclonal antibody, production of naked dsRNA by positive-strand RNA viruses, dsRNA viruses including reoviruses, and DNA viruses have been confirmed in infected cells[37]. It remains a puzzle, in the late phase of virus replication when the cellular viral dsRNA level is high and viral genome has obviously survived from cellular anti-viral immunity, whether the RNAi pathway is still functioning to silence non-viral reporter genes in the infected cells. In the present study, we designed an in vitro EGFP reporter system to monitor the RNAi effect in the context of GCRV replication. Results from this study could help clarify the mechanism of survival of reovirus genomic dsRNA from RNAi pathway of host cells.
HTML
-
Grass carp reovirus used in this study was the strain from CCTCC[30]. To propagate the virus, monolayers of grass carp Ctenopharyngon idellus kidney cells (CIK) were infected with crude extract of the infected kidney tissue or virus inoculum and incubated for 2-3 d at 28 ℃[38]. CIK cells were maintained in DMEM medium with 10% fetal bovine serum (Invitrogen). For the infection assay, Multiplicity of Infection(MOI) was optimized to be 1. Virus titration was as described as in a previous report[14].
-
Clarified supernatant from virus-infected cell cultures was subjected to ultracentrifugation. The pelleted viruses were re-suspended in 200 μmol /L E-MEM. For RNA extraction, all samples were processed with the QIAamp Viral RNA Mini Kit (QIAGEN) according to the manufacturer's protocol.
-
DIG-labeled RNA probes for egfp-dsRNA (egfp-probe) and GCRV genomic S10 fragment (S10-probe) were generated according to the protocol of the DiG RNA Labeling Kit (Roche). RNA probes were labeled with DIG-11-UTP by in vitro transcription with T7 RNA Polymerases. The template for the egfp-probe was a 730 bp PCR product containing full EGFP ORF with T7 RNA polymerase binding sites at both 5' ends. The fragment was amplified from pEGFP-N1 plasmid (Clontech) with primer pairs of 5'-GAATTAATACGACTCAC TATAGGGAGAATGGTGAGCAAGGGCGA-3' and 5'-GAATTAATACGACTCACTATAGGGAGATTACTTGTACAGCTCGT-3'. To prepare the template for the S10-probe, synthesis of the viral genomic cDNA was carried out in a reverse transcriptase reaction mixture (TAKARA) from purified viral genomic dsRNA with random primers. The cDNA was then used directly for PCR amplification of the S10-encoded vp7 gene fragment using the Master PCR system (Takara) with primer pairs of 5'-GAATT AATACGACTCACTATAGGGAGAATGCCACTTCACATGATTCCG'-3 and 5'-GAATTAATACGACTC ACTATAGGGAGACCAATCGGATGGCTCCAC-3'.
The resulting 930 bp vp7 ORF was flanked with T7 polymerase site at both ends and used for S10-probe synthesis through T7 transcription. Synthesized RNA probes were purified according to protocols of the supplier (Ambion), and unincorporated nucleotides were removed with G50 Sephadex Quick-Spin columns (Roche).
-
EGFP-specific dsRNA (egfp-dsRNA) was synthesized using a RiboMAX™ Large Scale RNA Production System-T7 (Promega) with the same 730 bp template as used in the synthesis of the EGFP-Probe. The concentration of the generated dsRNA was measured by the absorbance at 260 nm in a spectrophotometer. Duplex siRNA containing sequences positioned within the open reading frame of EGFP gene (egfp-siRNA: 5'-GAUGAACUUCAGGGUCAGCUU and UUCU ACUUGAAGUCCCAGUCG-5')[6] was obtained from Takara Biotech. The siRNA was synthesized with 3'-dTdT overhangs. The generated siRNAs were assessed by RNA gel electrophoresis on 2% agarose in TBE as described by the manufacturer. Typically in RNA transfection experiments, a 200 μL volume of Opti-MEM Ⅰ (Invitrogen) containing 2% Lipofectamine 2000 (Invitrogen) and 0.3 μmol/L duplex siRNA or 1 μg dsRNA was added to the medium overlaying each of the 12-well CIK monolayers. After incubation for 4 h, 1 mL of E-MEM containing 20% fetal bovine serum was added to each of the monolayers. Incubation was then continued for 24 to 48 h at 28 ℃.
-
For co-transfection of plasmid pEGFP-Nl and egfp-dsRNA or egfp-siRNA, 1 μg plasmid and 1 μL 30 μmol/L egfp-siRNA or 1 μg egfp-dsRNA was formulated into Lipofectamine 2000 (Invitrogen) with a final volume 0.4 mL/well. 1 μg plasmid pEGFP-Nl alone formulated into Lipofectamine 2000 was transfected separately as control. The cells transfected with pEGFP-N1 plasmid were subjected for inverted fluorescence microscopy analysis (Olympus) to monitor the gene expression of EGFP. The images were captured by a digital imaging system (Nikon). The fluorescence signal is a direct indicator of the functionality of RNAi pathway.
-
The transcriptional level of tested mRNA was determined by real-time quantitative PCR (qRT-PCR). The CIK cells (3 × 105) were cultured in 24-well plates at 28 ℃ for 24 h to a cell density of 70%~90%, then the cells were subjected for transient transfection or infection with GCRV. Total RNA was extracted using Trizol Reagent (Invitrogen) at different time points after transfection or infection. The RNA was resuspended in DEPC-treated water and stored at -80 ℃ until use. Reverse transcription of 1 μg total RNA was carried out with random hexamers in a total volume of 25 μL using a reverse transcription kit (Takara). cDNA was then used as template for qRT-PCR using a Two-Step qRT-PCR system (Promega). The housekeeping gene β-actin was measured in each sample and utilized as an internal control[36]. The forward and the reverse primers for the EGFP gene were: F-5'ACCAGCAGAACACC-3' and R-5'CCAGCAGGACCAT-3'. The primer sequences for the Dicer gene were: F-5'AGCCTTGTTACCTT TA-3' and R-5'TGGTTTAGCTGTTAGA-3'. The primer sequences for the housekeeping gene β-actin were: forward 5'-ATGTTGGTGACGAG-3' and reverse 5'-CCTCTGTGAGCAG-3'[31]. The EGFP replicon was 120 bp, the Dicer replicon was 131 bp and the β-actin product was 164 bp. After an initiating step at 95 ℃ for 2 min, the PCR mix was subjected to 40 cycles consisting of denaturation at 95℃ for 15 s and annealing and elongation at 60℃ for 1 min. Reactions were run in triplicate with iQ5 Detection System (Bio-Rad). The difference in the CT value between the tested gene and the corresponding internal control β-actin gene, ΔCT(CTβ-actin-CTEGFP), were calculated for each sample. The relative expression level of the target gene to β-actin was estimated using the equation 2−ΔCT and scaled by 100 to simplify data presentation.
-
Total RNA was isolated at indicated time points after transfection using 1 mL TRIzol reagent (Invitrogen) from each 6-well plate, and 15-20 μg purified RNA samples were run on a 1.2% agarose gel containing 2% formaldehyde. Small molecular RNAs were determined by molecular weight as indicated by the dsRNA Marker (Takara) through dye-staining before transferring the membrane. The separated RNA on the agarose gel was transferred to positively charged Nylon membranes (Bio-Rad) and UV-crosslinked. The membranes were pre-hybridized at 68℃ for 90 min and hybridized with the DIG-labeled RNA probes at 68 ℃ for 6 h according to the protocol of the DIG Northern Starter kit (Roche). After stringent washing, the membrane was sealed in plastic bags for chemiluminescent detection and imaging with a ChemiDoc XRS system (Bio-rad).
-
The presented data were representative of at least three experiments done in triplicate. The data were subjected to one-way analysis of variance (one-way ANOVA), followed by an unpaired, two-tailed t-test. A p value of less than 0.05 was considered as statistically significant.
Viruses and cell line
Preparation of viral genomic dsRNA
Synthesis of RNA probes
siRNA and dsRNA synthesis and RNA transfection
RNAi assay
RNA isolation, reverse transcription and quantification
Northern blot analysis
Statistics
-
The fact that RNAi mediated by GCRV-specific siRNA had been shown to inhibit virus infection in vitro suggested that there shouldn't be any GCRV-specific small molecular RNA during viral replication. In other words, the genomic dsRNA should be resistant to Dicer digestion to avoid the generation of GCRV-specific siRNA during viral replication. To verify this assumption, Northern blot analysis was performed to monitor the level of GCRV-specific small molecular RNA in CIK cells either infected with GCRV or transfected with purified GCRV genomic dsRNA. As a positive control, S10 fragment-specific small molecular RNAs were expected to be generated from cells transfected with S10-dsRNA due to Dicer digestion. Using a DIG-labeled S10-probe targeting the S10 fragment of the GCRV genome, small molecular RNA with homologous sequence to S10 was below the detectable level in the assay in total RNA samples prepared from CIK cells infected with GCRV (Fig. 1, lane 1); while, in cells without GCRV infection, both transfected GCRV genomic dsRNA and in vitro synthesized S10-dsRNA were degraded into viral sequence-specific small molecular RNA (Fig. 1, lanes 2 and 3). This result demonstrated that successful GCRV infection could protect its genome integrity from generating viral specific siRNA.
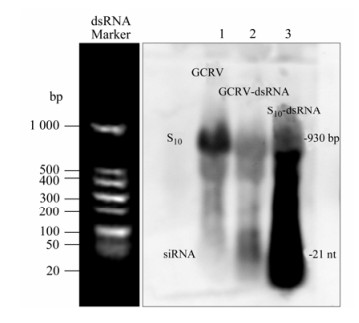
Figure 1. Northern blot analysis of GCRV-specific small molecular RNAs. RNAs were separated in gels of 1.2% agarose gel containing 2% formaldehyde. For the total cellular RNA sample, the amount of RNA analyzed was 15 μg (Lane 1); for the purified GCRV genome (Lane 2) and the in vitro synthesized S10-dsRNA (Lane 3), the amount of loading was 1 μg for each sample. DIG-labeled S10-Probe was used for hybridization. The dsRNA Marker (Takara) running with the RNA samples stained before gel-transferring was an indicator of the proper size of signals on the membrane.
-
Although siRNA-mediated gene silencing in CIK cells had been proven in previous report[20], failure to generate GCRV-specific siRNA might be due to the lack of cellular normal Dicer endonuclease activity toward dsRNA. To exclude this possibility, in vitro synthesized EGFP-dsRNA and pEGFP-N1 plasmid (1:1 ratio) were co-transfected into CIK cells. The gene silencing effects were monitored through inverted fluorescent microscopy on EGFP protein expression, Real time qRT-PCR analysis on EGFP mRNA transcription, and Northern blot assay on the siRNA level from the degradation of input egfp-dsRNA at 48 h post transfection. As a negative control, non-relevent S10-dsRNA and pEGFP-N1 (1:1 ratio) were co-transfected into CIK cells, which demonstrated strong fluorescent signal (Fig. 2A), high transcriptional level of the EGFP gene (Fig. 2B), and strong EGFP mRNA signal together with non-detectable level of EGFP-specific small molecular RNA in Northern blot assay (Fig. 3, Lane 1). In contrast, co-transfection of pEGFP-N1 with EGFP-dsRNA resulted in significantly reduced levels of fluorescent signal in transfected CIK cells (Fig. 2A), a 100 fold decrease in EGFP mRNA quantification (Fig. 2B), and degradation of both input egfp-dsRNA and EGFP mRNA into EGFP-specific small molecular RNAs (Fig. 3, Lane 2). These results supported the idea that the dsRNA-triggered and siRNA-mediated RNAi pathway was present in CIK cells. Thus it is possible to make further analysis on the interference of RNAi pathway by GCRV replication.
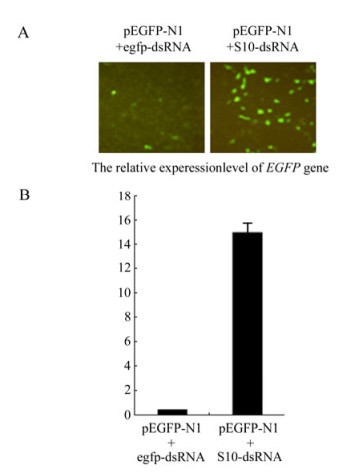
Figure 2. dsRNA-induced gene silencing in CIK cells. A: Fluorescence analysis of CIK cells co-transfected with pEGFP-N1 and synthetic egfp-dsRNA. At 48 h post transfection, cells were analyzed under inverted fluorescent microscope for EGFP expression. B: qRT-PCR analysis of the mRNA level of EGFP gene in cells co-transfected with pEGFP-N1 and the synthetic egfp-dsRNA. Each column represents the level of EGFP mRNA relative to β-actin RNA as described in section of Materials and Methods. Error bars represent the calculated standard error of measurement (SEM). Cells co-transfected with pEGFP-N1 and S10-dsRNA served as negative control for both A and B.
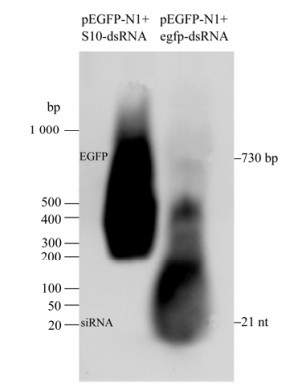
Figure 3. Northern gel analysis of EGFP-specific small molecular RNA. RNAs were separated in gels of 1.2% agarose gel containing 2% formaldehyde. For the total cellular RNA sample from transfected cells, the amount of RNA analyzed was 15 μg. DIG-labeled EGFP-Probe was used for hybridization. The dsRNA Marker (Takara) run with the RNA samples stained before transferring to the nylon membrane was an indicator of the proper size of signals on the membrane.
-
We designed an infection and transfection experiment to test the effect of GCRV replication on the RNAi pathway in CIK cells. CIK cells were infected with GCRV at a MOI of 1, and then pEGFP-N1 & the synthetic EGFP-siRNA (1:1 ratio) were co-transfected into the cells at 4 h post infection. pEGFP-N1 alone was transfected into GCRV-infected CIK cells as a negative control and mock-infected CIK cells transfected with pEGFP-N1 & EGFP-siRNA served as positive controls for the RNAi assay. The gene silencing effect was directly reflected by inverted fluorescent microscopy at 24 h post transfection. Fig. 4A shows that in contrast to the mock-infected CIK cells, GCRV-infected CIK cells transfected with pEGFP-N1 & EGFP-siRNA demonstrated a strong fluorescent signal similar to those cells transfected with pEGFP-N1 only. The transcriptional levels of EGFP gene were further determined by real-time qRT-PCR of total cellular RNA samples to validate the RNAi effect. The amount of EGFP mRNA in GCRV-infected cells transfected with pEGFP-N1 & EGFP-siRNA was found to be similar to those infected cells transfected with pEGFP-N1 only, both of which were 10 fold higher than mock-infected cells transfected with pEGFP-N1 & egfp-siRNA(P < 0.05)(Fig. 4B). These results indicated that siRNA-mediated RNAi was inhibited by GCRV replication in CIK cells.
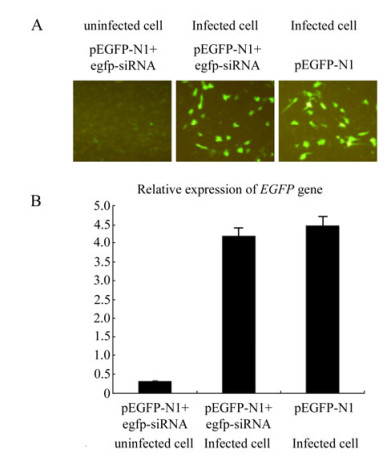
Figure 4. Infection-transfection experiments to assess the effect of GCRV replication on RNAi. A: Fluorescence analysis of mock-infected and GCRV-infected CIK cells co-transfected with pEGFP-N1 and EGFP-siRNA. Infected CIK cells transfected with pEGFP-N1 served as negative control. At 24 h post transfection, cells were analyzed under inverted fluorescent microscope for EGFP expression. B: qRT-PCR analysis of the mRNA level of EGFP gene in mock-infected and GCRV-infected CIK cells transfected with pEGFP-N1 & EGFP-siRNA, Each column represented the level of EGFP mRNA relative to β-actin RNA as described in section of Materials and Methods. Error bars represent the calculated standard error of measurement (SEM). GCRV-infected Cells transfected with pEGFP-N1 served as negative control for both A and B.
-
So far we have shown that no GCRV-specific small molecular RNA could be detected and in vitro synthesized siRNA-mediated gene silencing was inhibited in CIK cells infected with GCRV, suggesting that the loss of the function of the RNAi initiator protein Dicer in producing siRNA might not be the only factor accounting for the impaired RNAi pathway during viral replication. We continued to test the steady-state transcription of Dicer gene during viral replication in CIK cells. The infected cells were collected at 0, 4, 8, 12, 16, 20, 24, 28 h post infection for extracting total cellular RNA. qRT-PCR was performed to detect the Dicer gene expression. The expressions of the Dicer mRNA increased dramatically after 20 h (P < 0.05) post infection (Fig. 5A). Interestingly, the TCID50 titration assay revealed that viral replication approached the maximum level at 20 h post infection (Fig. 5B). Obviously, the successful viral genome replication was not correlated with the blocking of transcriptional levels of Dicer gene, and viral genome dsRNA in the late infection was still resistant to Dicer nuclease even when its transcriptional level was highly induced. Our results suggested that Dicer gene transcription positively responded to the replication level of GCRV. Given that purified GCRV genomic dsRNA was sensitive to Dicer digestion in uninfected cells and GCRV could comfortably coexist with the high transcriptional levels of Dicer gene in infected cells, some unidentified viral protein factors were thus expected to cause the loss-function of Dicer protein during viral replication.
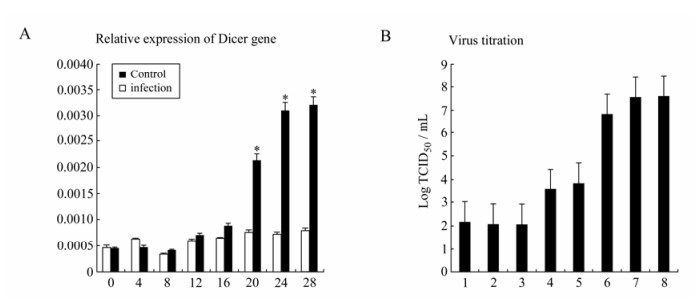
Figure 5. A: qRT-PCR analysis of state-steady expression level of Dicer mRNAs after GCRV infection in CIK cells. Each column represented the level of Dicer mRNA relative to β-actin RNA expressed as the mean±SE of triplicate qRT-PCR assays (2ΔCT) for three different cDNA samples. Asterisk indicates a significant difference between experiment group and control group with t-test (P < 0.05). B: The GCRV titration curve through TCID50 assay of the supernatants of GCRV-infected CIK cells. CIK cells were infected with a MOI of 1.
Infection of CIK cells with GCRV resulted in the protection of its dsRNA genome from degradation into small molecular RNAs
Transfected EGFP-dsRNA efficiently induce siRNA-mediated gene silencing through the accumulation of EGFP-siRNA in CIK cells
GCRV replication resulted in the inhibition of siRNA-mediated gene silencing on the reporter EGFP gene
Augmented Transcriptional level of Dicer gene is correlated with the replication level of GCRV in CIK cells.
-
The RNAi machineries of invertebrates and vertebrates share many common features. Besides mammalian cells, dsRNA-induced silencing has been harnessed as a powerful tool for the analysis of gene function in several systems including plants, fungi and metazoans[28]. Compared with other multicellular organisms, little is known about dsRNA-triggered RNAi pathway in fish cells, except for zebrafish[4, 34]. Employing northern blot analysis (Fig. 1 and 3), we were able to show that the dsRNA dominantly silences gene expression in a sequence-specific manner by causing the corresponding endogenous mRNA to be degraded in grass carp cells. It drove us to explain why the GCRV dsRNA genome could survive from the dsRNA-triggered and Dicer-initiated RNAi pathway in CIK cells.
A previous study implied that the failure of generating GCRV-specific siRNA might account for the successful replication of GCRV through a transfection and infection assay[20], in which GCRV specific siRNA was transfected into host cells followed by virus infection. Since the RNAi-initiator protein Dicer was responsible for the production of siRNA from digestion of RNAi-trigger dsRNA, inhibition of Dicer function by some unknown viral factors might be the major mechanism resulting in the failure of the activating RNAi pathway in CIK cells infected with GCRV. It is notable that our results were so far in consistent with this assumption. As well as the functional inhibition of Dicer protein, we found that GCRV replication inhibited siRNA-mediated gene silencing, through an infection and transfection assay, in which CIK cells were first infected with GCRV followed by co-transfection with EGFP-siRNA and the pEGFP-N1 plasmid (Fig. 4). We thus concluded that GCRV antagonizes the RNAi pathway in multiple steps. One advantage of this anti-RNAi strategy might be to offer comprehensive protection of the viral genome in case each single step was not 100% inhibited during viral replication. Indeed, we discovered that Dicer transcription responded positively to the dsRNA level of GCRV genome and up to 100 fold higher transcriptional levels were detected at 20 h post infection when the viral load was high (Fig. 5). It could be possible that miniscule quantities of uninhibited Dicer protein might be present when its transcriptional level was dramatically increased. Considering that GCRV genomic itself was sensitive to Dicer digestion, minute quantities of free Dicer protein would still pose a big threat to viral replication. Inhibition of RNAi pathway downstream of the Dicer initiator would diminish this threat to the minimum level.
It is the subject of some controversy as to whether RNAi acts as an antiviral immune system in vertebrates, similar to that established in plants and invertebrates. Production of virus-derived small RNA is undetectable in mammalian cells with RNA viruses[1]. We deduced from our results that the CIK cells reserve the antiviral machinery of conservative RNAi pathway (Fig. 2), although GCRV holds an efficient counter-defense strategy against RNAi that needs further characterization. The data here support the general idea that the RNAi pathway poses an innate viral immunity that responds immediately upon viral attack. However, more host proteins beyond Dicer are expected to bind to and be activated by dsRNA, including protein kinase R (PKR), oligo(A) synthetase, and adenosine deaminase[7]. A comprehensive investigation on the interaction between viral genomic dsRNA and all these host receptors would accurately clarify the role of RNAi pathway in anti-viral immunity.
In summary, the dsRNA genome of GCRV alone was sensitive to Dicer digestion, while efficient GCRV replication correlated with the inhibition of RNAi pathway in host cells. Successful replication of GCRV in CIK cells was not due to the blocking of transcription of the Dicer gene; on the contrary, we found viral replication could enhance its transcription. The resistance of GCRV genomic dsRNA to Dicer was not fully responsible for its immunity from RNAi pathway, and the inhibition of downstream effector nuclease of host RNAi pathway might play an equally important role for its survival in host cells. Further characterization of the interaction between viral proteins and host RNAi components would be necessary to understand the details on the survival of GCRV genome from RNAi pathway in CIK cells.


 DownLoad:
DownLoad: