









-
Lyssavirus is an unsegmented, single-strand, negative sense RNA virus belonging the Rhabdoviridae family. The lyssavirus genome is approximately 12 kb and comprises five genes (3'-5'), encoding nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G) and RNA-dependent RNA polymerase (L) (Nel L H, et al., 2007). Currently, there are twelve defined lyssavirus species on the basis of their genetic similarity and antigenic patterns (9th ICTV classifications: http://ictvonline.org/virusTaxonomy.asp?version=2011&bhcp=1).
Rabies virus (RABV) is the prototype and most studied virus of the lyssavirus genus and several whole genomes of the typical vaccine strains of RABV have been sequenced, i.e. PV, SAD B19, Nishigahara, RC-HL, HEPFlury, ERA and RV-97 (Conzelmann K K, et al., 1990; Geue L, et al., 2008; Inoue K, et al., 2003; Ito N, et al., 2001; Metlin A, et al., 2008; Tordo N, et al., 1986; Tordo N, et al., 1988); There are also genomic characterizations of street strains in RABV isolates from different countries and hosts (Gould A R, et al., 2002) as well as lyssavirus genomic analysis of other genotypes (Delmas O, et al., 2008; Faber M, et al., 2004; Horton D L, et al., 2010; Kuzmin I V, et al., 2010; Le Mercier P, et al., 1997; Mochizuki N, et al., 2009; Nagaraja T, et al., 2008; Szanto A G, et al., 2008; Warrilow D, et al., 2002).
Rabies is an acute encephalomyelitis, with its symptoms including a variety of neurological disorders brought about by the viral interference in the central nervous system (Nel L H, et al., 2007). Rabies has been endemic in China for a long time, and more than 2000 cases have been reported every year since 2001 (Song M, et al., 2009). To better understand the genetic features of rabies virus strains circulating in China, the genomes of a human vaccine strain(CTN181) and a several street strains in China were sequenced and analyzed (Du J, et al., 2008; Ming P, et al., 2009).
In this study, we report the full-length genomic sequence analysis of the aG strain, the first human rabies virus vaccine strain in China. Although there has been a previous report on this aG strain (Jiao W, et al., 2011), this was a relatively straightforward comparison against genome sequences at the nucleotide and amino acid level. In this work we present a more comprehensive analysis of the aG strain genome sequence relative to other sequenced street lyssavirus genomes as well as the other typical vaccine strain genomes available from GenBank.
It is generally considered that the G protein plays a predominant role in determining the pathogenicity of the virus (Faber M, et al., 2004). Hence, to explore whether the difference in the pathogenicity of vaccine and street strains can be associated with the limited amino acid mutations on the G sequences, we predicted the tertiary structure of the G protein of the aG strain, CTN181 strain and wild type strain HN10, based on the crystal structure of Vesicular stomatitis virus (VSV) G (Roche S, et al., 2006). By mapping the characterized mutations to this structure we attempted to analyze the possible molecular basis of the differences in virulence between the RABV vaccine strains and street strains.
HTML
-
The parental virus of aG was isolated from a rabid dog brain from Beijing in China in 1931. After about 30 passages in rabbit brain, the street strain then became fixed. After another 55 passages in primary hamster kidney cells (PHKC), followed by 3 passages alternatively in brains of guinea pigs and PHKC, the fixed strain was named aG strain. A rabies vaccine using aG strain passaged in primary hamster kidney cells was successfully developed and certified for production in 1980s in China.
-
Total RNA was extracted with TRIzol Reagent (Invitrogen, Carlsbad, CA, USA), and then used as the template for cDNA synthesis with Ready-To-Go You-Prime First-Strand Beads (Amersham Pharmacia Bioscience, Chalfont St.Giles, Bucks, UK). The fragments of the genome were amplified, and the 3'-terminal end and 5'-terminal end were confirmed using a RLM-3' RACE and 5'RACE kit (Invitrogen), with a similar method as described previously (Zhang J, et al., 2012). The PCR products were purified with an QIAquick PCR Purification Kit (Qiagen Ltd., Crawley, West Sussex, UK), then sequenced with an ABI PRISM 3100 DNA Sequencer.
-
The lyssaviruses used in this study are shown in Fig. 1. For the street lyssaviruses, all available sequences from GenBank were included (as of Jan 6, 2011), whereas for the rabies vaccine strains, only representative sequences were selected. Multiple alignments for whole genomes, and for the alignment of concatenated coding regions of the N, P, M, G and L genes, were produced using the ClustalX v2 program (Thompson J D, et al., 1997). The identity values were calculated with the MegAlign software package (v5, DNAStar, Inc., Madison, WI, USA). Phylogenetic analysis was performed using the Neighbor joining (NJ) method (Roche S, et al., 2006) implemented in the MEGA 5 program with 1000 bootstrap replicates (Kumar S, et al., 2004).
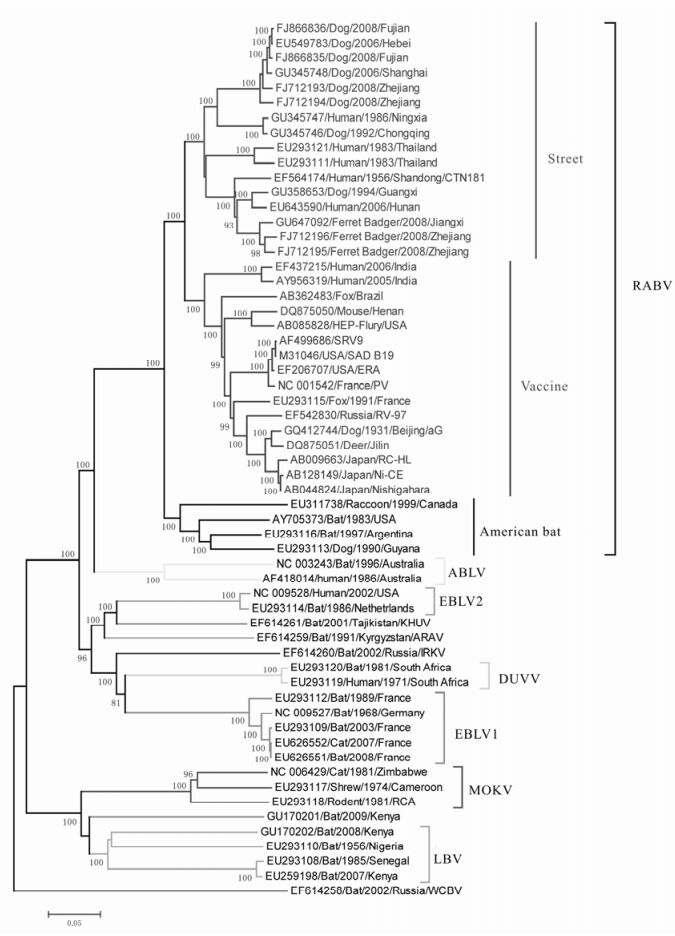
Figure 1. NJ tree of 59 genomes of lyssavirus. In RABV, blue lines indicate street strains, and red lines indicates vaccine strains.
-
The 3D structure of the G protein of RABV was predicted based on homology crystal modeling of VSV (Roche S, et al., 2006 (Roche, 2006), which is another genus of Rhabdoviridae family. Using the VSV G structure 2J6J as a template, an initial model of RABV G was generated using Modeller (http://salilab.org/ modeller/) (Sali A, et al., 1995).
Virus strain
Sequence determination
Sequence analysis
Protein Structure Modeling
-
The complete aG genome was 11925 nucleotides in length (GenBank accession no. GQ412744), similar to strains D01, D02 and 8764THA. The genomic organization of aG, which is typical of all previously characterized RABVs, can be summarized as follows: a 3' leader region of 58 nucleotides (1-58), the N gene (59-1482), P gene (1485-2476), M gene (2482-3284), G gene (3290-5356), L gene (5381-11855), and the 5' trailer region of 70 nucleotides (11856-11925). The coding regions of the five structural genes are manifested as follows: 1353nt N (71-1423), 894nt P (1515-2408), 609nt M (2497-3105), 1575nt G (3317-4891) and 6384nt L (5411-11794).
-
For the 59 genomes listed in Fig. 1, the size of coding regions, non-coding regions and whole genome were calculated respectively and are shown in Table 1. All genomes have the same structural organization although their lengths vary between 11918nt (ABLV) and 12278nt (WCBV). The 3'UTR is invariably conserved in length (70nt), while the length of other non-coding regions are variable. The predicted size of the coding regions is similar among genotypes, with the M protein identical in length across all genotypes and the P protein the most variable (894-918nt).
LYSSAs 3'UTR N N-P P P-M M M-G G G-L L 5'UTR Genome Rabies virus (RABV) 70 1353 73-91 894-906 76-88 609 211-215 1575 515-527 6384-6429 86-131 11908-11932 Lagos bat virus (LBV) 70 1353 101-103 918 75-76 609 204 1569 574-588 6384 143-146 12003-12017 Mokola virus (MOKV) 70 1353 100-102 912 80-81 609 203 1569 546-562 6384 112-114 11940-11957 Duvenhage virus (DUVV) 70 1356 90 897 83 609 191 1602 562-563 6384 131 11975-11976 European bat Lyssavirus 1
(EBLV1)70 1356 90-96 897 83 609 211 1575 560 6384 130-131 11966-11971 European bat Lyssavirus 2
(ELBV2)70 1356 101 894 88 609 205-210 1575 511-512 6384 131 11924-11930 Australian bat Lyssavirus
(ABLV)70 1353 93-94 894 87 609 207-209 1578-1581 508-509 6384-6387 103-131 11918 Khujand virus (KHUV) 70 1356 95 894 72 609 208 1581 504 6384 130 11903 Irkut virus (IRKV) 70 1356 92 897 83 609 214 1575 569 6384 131 11980 Aravan virus (ARAV) 70 1356 85 894 85 609 210 1581 514 6384 130 11918 West Caucasion bat virus
(WCBV)70 1353 64 894 133 609 206 1578 862 6384 125 12278 Shimoni bat virus
(SHIBV)70 1353 98 918 76 609 205 1569 613 6384 150 12045 Table 1. Coding regions, non-coding regions and genome size (in nucleotides) of the twelve different species in the lyssavirus genus.
-
To estimate the evolutionary relationships among the lyssaviruses we performed a phylogenetic analysis utilizing the whole genome of 59 strains representative of the twelve lyssavirus species. The predicted tree is shown in Fig. 1 and reveals the separation of lyssaviruses into 3 major branches: the first major branch includes Rabies KHUV, ARAV and IRKV comprising phylogroup 1; the second major branch contains LBV, MOKV and SHIBV denoted as phylogroup 2; while the single WCBV belongs to phylogroup 3 (Nel L H, et al., 2007).
All strains from China cluster together within RABV which could be further divided into 3 subclades. The first subclade (Fig. 1, Street) mainly contains Chinese street strains that were isolated in recent years, but also includes CTN181, the cloned strain of vaccine strain CTN (Du J, et al., 2008). The second subclade (Fig. 1, Vaccine), includes the standard world vaccine strains, street strains from India, Brazil, France and China, and the Chinese vaccine strain aG (Tao X Y, et al., 2009). The American bat strains and their spillover strains comprise the third subclade (Fig. 1, American bat) (Badrane H, et al., 2001). Overall, the phylogenetic tree indicates that the aG strain isolated earlier has a closer relationship with the world vaccine strains whereas the CTN181 strain that was isolated later clusters with the street strains that have been epidemic in recent years.
-
The G protein is responsible for cell attachment and fusion and is the main viral protein responsible for the induction of neutralizing antibodies and cell-mediated immune responses (Thoulouze M I, et al., 1998; Wunner W H, et al., 1987). It plays a predominant role in viral pathogenicity (Faber M, et al., 2004).
In this study, the 3D structure of the ectodomain of the RABV G protein was predicted (Fig. 2) based on the solved crystal structure of the VSV G protein (Roche S, et al., 2006). The predicted structure is consistent with the VSV G protein and the ectodomain of RABV G can be similarly divided to 4 distinct domains (Fig. 3): Domain (D) Ⅰ is completely composed of β sheets, and is constructed from two separate regions; DⅡ mainly consists of α helices formed from 3 sequences segments; DⅢ involves multiple β sheets and a few α helices, and is formed from two sequence segments; DIV comprises β sheets and α helices, forming a continuous sequence.
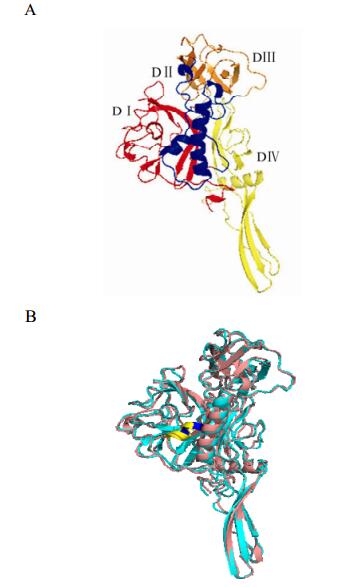
Figure 2. A: The overall predicted structure of the G ectodomain of RABV for vaccine strain aG. The colours red, blue, orange and yellow correspond to the VSV G protein domains DI-DIV respectively. B: superimposed predicted structures for CTN181 and wild type strain HN10. The two structures show high similarity except for the secondary structure in the region of site 303. In the vaccine strain, the mutation is predicted to form a short α-helix (marked in yellow and blue) which is longer in the wild type strain.
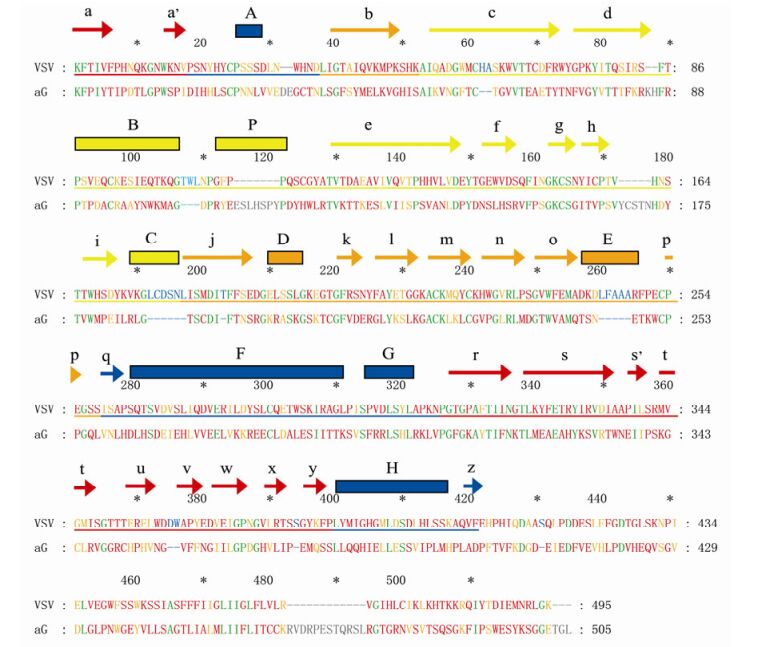
Figure 3. Alignment of amino acid (aa) sequences of the VSV and aG strain G protein. The secondary structure are marked above the sequences and colour-coded corresponding to the domains in Figure 2a.The arrows indicate β sheets, and the rectangles indicate α helixes. Green letters indicate identical residues, orange letters indicate similar residues, red letters indicate sequence mismatch, blue letters indicate deletion, and grey letters indicate insertion.
-
HN10 is a street strain that was isolated in Hunan province in China and has the highest sequence identity with CTN181; only 17 mutations were found within the 505 amino acid (aa) sequences of the mature G proteins, 13 of these mutations were located in the ectodomain sequence (Fig. 4). For the variable sites located in the ectodomain, most of the amino acid substitutions are of similar nature (Venn diagram of amino acid classification, http://blog.bioon.net/user1/2081/archives/2008/173490.shtml). However, at site G303, the two strains have different amino acids with distinct properties: for HN10 strain the amino acid is H (Histidine) and the site is within an α helix (298-304); for CTN181 strain is P (Proline), and 301-304 sites constructed a α helix (shorter than HN10) (Fig. 2b). Therefore, the mutation H303P appears to produce a change in spatial structure of DⅡ. These differences are also present in other wild strains at site 303(Fig. 5). We therefore speculate that the G303 mutations may well be associated with the virulence difference between the vaccine and wild type strains.

Figure 4. Alignment of amino acid sequences of HN10 and CTN181 strains G.

Figure 5. Alignment of amino acid sequences of CTN181 and other street strains G. Only CTN181 is P for G303 site, while other street strains are H or Y for this site.
By comparing the G protein amino acid sequences of the aG strain and other 14 strains (Fig. 1, Vaccine clade, except DRV strain), we found there were two specific mutations in the aG strain (Fig. 6): site G165, within a β sheet in DIV, is proline (P) in aG, but serine (S) for the other 14 strains; the G231 site is located within a β sheet in DⅢ and is P for aG, but Leucine (L) for the others strains. Since proline cannot form a hydrogen bond between the β sheets, the direction of the β sheet may be modified and so these two particular mutations may influence the connectivity and hence the conformation of the secondary structure.

Figure 6. Alignment of amino acid sequences of aG and other 14 strains G..
Sequence features of aG genome
Genome comparisons
Whole genome phylogenetic analysis of lyssaviruses
G protein structure modeling
Comparison of Chinese rabies vaccine strains with street strains
-
Until now, only two Chinese strain aG and CTN have been approved as seed strains for vaccine production by WHO and the corresponding organizations in China (Du J, et al., 2008). The aG strain was the first attenuated RABV strain in China, and was also the first Chinese isolated strain used for vaccine production in China. Some structural genes of aG have already been sequenced and reported by our lab (Du J, et al., 2008). In this study, we report the complete genome sequencing of aG. Another Chinese vaccine strain's cloning strain, CTN181, have been reported by our lab previously (Du J, et al., 2008).
Analyses of both the whole genome and single gene sequences indicated that there are clear distinctions between the aG and CTN181 strains with aG showing a close relationship to the classical vaccine strains worldwide but CTN181 demonstrating more similarity to epidemic street strains predominant in China (Fig. 1). The differences may be a consequence of the difference between the isolation location and isolation date of the two strains. Regardless, both of them are equally effective for rabies prevention.
Regarding the differences between vaccine strains and street strains of rabies viruses, many comparison and analysis have reported and tried to explain the possible reasons for their differing virulence (Du J, et al., 2008; Meng S L, et al., 2007; Ming P, et al., 2009; Tao X Y, et al., 2009). In this work, using sequence analysis and 3D protein structure modeling, we searched for specific genetic traits that might contributes to the viral virulence in order to understand possible reason for attenuation of virulence. It has been demonstrated that amino acid composition and consequent modification of spatial structure can affect protein function (Desmezieres E, et al., 2003; Gao Y, et al., 1996; Gaudin Y, et al., 1992; Roche S, et al., 2002). The G protein plays an important role in RABV pathogenicity (Faber M, et al., 2004). Therefore we investigated changes that might impact the structure of the G protein. The CTN181 strain shows the most similarity with street strain HN10, but has 13 mutations within the G ectodomain. Among these changes, the G303 mutation appears significance as it affects the formation of an α helix within the DⅡ domain. The aG strain has two signature mutations (G165P and G231P) when comparing the G protein sequence with other 14 closely related street strains. Both these sites are located within β sheets, suggesting that they may both also be of structural importance and sassociated with function and hence attenuation of virulence.


 DownLoad:
DownLoad: