









-
Human cytomegalovirus (HCMV), a member of the Betaherpesvirinae subfamily, is a ubiquitous pathogen that infects approximately 60 to 80% of the adult population worldwide (Boeckh M, et al, 2011). It generally causes asymptomatic latent infection in immunocompetent populations, but leads to serious illness and even death among immunocompromised populations, such as transplant recipients, infants with an immature immune system, and patients infected with human immunodeficiency virus (HIV) (Lazzarotto T, et al, 2011; Sung H, et al, 2010). Thus, HCMV infection is an important public-health problem. There are some anti-viral drugs currently used to treat HCMV infection in clinical practice, but their curative rates are not satisfactory, and there are issues such as severe side effects and emergence of drug-resistant HCMV strains (Griffiths PD, 2002; Manley K, et al., 2011). The most effective preventive measure against HCMV infection will therefore be vaccination; unfortunately, more than 30 years of research efforts have not yielded a usable HCMV vaccine (Schleiss MR, et al, 2005). Hence, the development of a CMV vaccine has been assigned the highest priority by the US Institute of Medicine (Arvin AM, et al, 2004; Zhong J, et al, 2007).
HCMV has broad cell tropism, and the pathway for HCMV entry into cells is very complex, involving interactions of multiple viral envelope glycoproteins and a series of cell receptors (Britt WJ, et al, 1996). The HCMV envelope mainly contains three glycoprotein complexes (gC Ⅰ, Ⅱ, and Ⅲ), which are all necessary for virus infection (Wang D, et al, 2005). Of these, the gC Ⅱ complex, consisting of the gM (also known as gpUL100) and gN (gpUL73) proteins, is the most abundant glycoprotein complex. The gM protein accounts for 10% of the net weight of the virus, and studies have shown that knockout of either the gM or gN genes is a lethal mutation (Britt WJ, et al., 2004; Krzyzaniak M, et al., 2007). The gC Ⅱ complex binds to the heparan sulfate proteoglycan and is inferred to play a role in the initial steps of virus entry into cells. In addition, the gM protein also participates in virus replication and assembly (Krzyzaniak M et al, 2007), and the carboxyl terminal of the gN protein plays a key role in virus envelopment (Mach M, et al, 2007). It has been reported that natural HCMV infection can induce antibody responses against the gC Ⅱ complex (Mach M, et al, 2000; Shimamura M et al, 2006), and that monoclonal antibodies against gM or gN are capable of neutralizing HCMV infection, indicating that, similar to the gB and gH proteins, the gM and gN proteins are also major target antigens in anti-viral immune responses.
Currently, HCMV vaccine development is focusing mainly on the gC I antigen gB, and the related vaccine has entered clinical Phase Ⅲ trials. However, clinical studies have shown that the protection offered by the gB protein is only about 50% (Pass RF, et al, 2009), therefore, it is necessary for research and development to be carried out into new antigens that give greater protection. As not only is the gM/gN complex highly conserved in the Herpesviridae family and capable of inducing neutralizing antibody (NAb) response, but it is also the most abundant glycoprotein of HCMV and has become an attractive candidate vaccine antigen. Shen et al. reported that HCMV gM and gN DNA vaccines induced neutralizing antibodies that, in in vitro tests, demonstrated neutralizing activity against multiple HCMV strains (Shen S, et al., 2007). However, as CMV infection has strict species specificity, no animal model is available for studying the mechanisms of HCMV infection and immunity, and so far no in vivo protective study using gM-gN as vaccine antigens has been reported. Mice infected with murine cytomegalovirus (MCMV) has been the most commonly used animal model for simulating HCMV infection (Brune W, et al, 2001; Qureshi, MH, et al, 2005). So far there have been few reports describing the gM/gN proteins of MCMV. The M100 and M73 open reading frames of the MCMV genome encode the homologs of gM and gN, respectively. MCMV gM is speculated to comprise eight transmembrane domains and four N-linked glycosylation sites. Anthony et al. found that MCMV gM was completely conserved between six MCMV strains, suggesting that gM is an antigenically conserved protein (Scalzo A A, et al., 1995).
Generation of a DNA vaccine has long been an important direction for CMV vaccine research and development. Currently, a number of DNA vaccines have entered into clinical trials, such as VCL-CB01 (Phase Ⅱ) and VCL -CT02 (Phase Ⅰ) (both Vical Inc., San Diego, CA, USA), in which the protective antigens selected are gB/pp65 and gB/pp65/IE1, respectively. In this study, we prepared gM and gN DNA vaccines based on sequences of the MCMV Smith strain, and the two DNA vaccines were then tested separately and in combination in a lethal MCMV infection mouse model. The results showed that gM-gN antigens have good immunogenicity, and co-immunization with gM and gN DNA vaccines was able to provide the mice with complete protection against a lethal MCMV infection.
HTML
-
The MCMV Smith strain was used and propagated in NIH 3T3 cells. MCMV from such cell-culture propagation is referred to as tissue culture-derived MCMV (TC-MCMV). MCMV isolated from mouse salivary glands (SG) followed by in vivo passage for virulence enhancement is referred to as salivary gland (SG)-MCMV, and in this study was used in challenge experiments. The highvirulence SG-MCMV stock had a 50% lethal dose (LD50) of approximately 105 plaque-forming units (PFU) in BALB/c mice, and challenge was performed with 5× LD50 virus stock.
Female BALB/c mice were purchased from the Center for Disease Control in Hubei Province, China, and kept under specific-pathogen-free conditions in the Animal Resource Center at Wuhan Institute of Virology, CAS. All procedures were inspected and ratified by the Animal Care Committee of the Wuhan Institute of Virology.
-
DNA vaccines were constructed by cloning the complete open reading frames of gM and gN genes from the MCMV Smith strain into the plasmid expression vector pcDNA3.1. We also constructed a gB DNA vaccine, which encoded only the extracellular domain of gB. All constructs were sequenced in full. The plasmids were cultivated in Escherichia coli DH5α bacteria and purified using NucleoBond® Xtra kits (MACHEREY-NAGEL GmbH & Co. KG, Düren, Germany).
-
The 293T cells were seeded into six-well plates, then 24 hours after plating, cells were transfected with the vector plasmid, plasmid with gM or gN (pgM or pgN), or co-transfected with pgM/pgN using a commercial reagent (Lipofectamine 2000; Invitrogen Corp., Carlsbad, CA, USA) in accordance with the manufacturer's instructions. The cells were harvested and lysed 48 hours after transfection. For the infection experiment, 3T3 confluent cells were infected with MCMV (multiplicity of infection (MOI) = 1) for 1.5 hours at 37℃. Unabsorbed virions were removed and infection medium was added. Cells were collected at 3 days post-infection, and subsequently lysed. Only the lysates from the pgM/pgN co-transfected 293T cells and MCMV-infected 3T3 cells were prepared in non-reducing sample buffer. Cell lysates were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 10% gel), blotted onto polyvinylidene difluoride (PVDF) membranes and immunoblotted with polyclonal antisera to gM or gN. Antibody binding was detected using horseradish peroxidase (HRP)-labeled goat anti-mouse IgG and an enhanced chemiluminescence detection system.
-
BALB/c mice (female, 6-8 weeks old) were randomly divided into seven groups. Groups A, B, and C were immunized three times, 3 weeks apart, using 50 μg of pgB, pgM, and pgN (referred to as 3×pgB50, 3×pgM50, and 3×pgN50, respectively), with 3×pgB50 being the positive control. Groups D, E, and F received co-immunization with pgM and pgN two or three times with a 3-week interval: group D received 50 μg pgM + 50 μg pgN three times (3×pgM/pgN50); group E received 50 μg pgM + 50 μg pgN two times (2×pgM/pgN50); and group F received 25 μg pgM + 25 μg pgN three times (3×pgM/pgN25). Group G served as the negative control, and received the empty DNA vector (3×pV50) three times. The mice were vaccinated by inoculation into the right quadriceps muscle with the corresponding DNA vaccines. In vivo electroporation was then carried out according to a previously described method (Aihara H, et al., 1998). First, a pair of electrode needles were inserted into the muscle to cover the inoculation sites, then electric pulses were delivered using an electric-pulse generator (Electro Square Porator T830 M; BTX, San Diego, CA, USA). Three pulses of 100 V, each sustained for 50 ms, were administered to the inoculation site, followed by three pulses of the reverse polarity.
For lethal dose experiments, the more virulent SGMCMV was used. At 3 weeks after vaccination, mice were infected with a lethal dose (5×LD50, 200 μL/mouse) of SG-MCMV by intraperitoneal injection. This infection was able to cause systemic virus replication in mice and death of all unvaccinated mice within 1 week after infection. During the 3 weeks following the virus challenge, the body weight of each mouse was recorded until death occurred.
-
Mouse serum samples were collected 2 weeks after immunization and stored at -20 ℃ until use. NAbs directed against MCMV were measured by plaque reduction assay as described previously(Geoffroy F, et al., 1996). Serum samples were diluted twofold serially. Neutralization titers were presented as the highest dilution of sera required to achieve a 50% reduction in the number of plaques.
-
3T3 cells grown on glass coverslips were infected with MCMV (MOI = 0.1) for 1.5 h at 37℃, then fresh medium was added. Three days later, the cells were washed with phosphate-buffered saline (PBS), fixed in paraformaldehyde and permeabilized with 0.5% Triton X-100 in PBS. A blocking process was performed with 5% non-fat dry milk, then antiserum to gM/gN was used for the reaction. Secondary antibody (fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG; Millipore Corp., Bedford, MA, USA) was added, and sections were then stained with Hoechst dye. Fluorescent images were captured on a laser scanning confocal microscope (Leica, Heerbrugg, Switzerland) as described previously (Chen J J, et al., 2010).
-
At 5 days after challenge, three mice from each group were euthanased and the spleens removed aseptically for measurement of viral titer. The remaining mice were recorded in the survival and weight-loss data records. Spleens were homogenized in 1:10 (w/v) volume of minimal essential media containing 10% calf serum. The homogenized fluids were centrifuged and the supernatants stored in aliquots at -80℃.
Viral loads were determined using a plaque-forming cell assay. Briefly, organ homogenates were serially diluted 10-fold, and each dilution was used to infect 3T3 cells cultured in 48-well plates. Infections were performed in triplicate, and 100 μL viral dilution was added to each well. After 1.5 h of absorption, the supernatant was aspirated, then 0.5 mL viscous medium was added to each well. After incubation for 4 to 6 days, viral plaques were counted and the viral PFU/mL calculated. The viral titer of each group was defined as the mean ± SD of the viral t iter per ml of the mouse samples in each group.
-
Mice were vaccinated three times with pgM, pgN or the pgM/pgN combination at a dose of 50 μg. Two weeks after the third immunization, splenocytes were collected for ELISPOT assays. Following the manufacturer's instructions (U-CyTech, Netherlands), immunospot plates (Millipore Corp., Bedford, MA, USA) were coated with rat anti-mouse interferon (IFN)-γ monoclonal antibody (mAb), cultivated at 4 C overnight, and then blocked with 200 µL of blocking liquid R. Subsequently, 2 × 105 splenocytes were seeded in triplicate wells, and stimulated with 10 µg/mL of formalin-inactivated (FI)-MCMV. After 18 h, splenocytes were discarded and biotin-labeled anti-mouse IFN-γ Ab was added. Next, diluted liquid streptavidinHRP conjugate was added and the plates incubated for 2 hours. Finally, 100 µL of 3-amino-9-ethylcarbazole (AEC) substrate was added and the plates incubated for 20 min in the dark. Spots were quantified by an ELISPOT reader (Bioreader 4000; BIO-SYS GmBH, Karben, Germany).
-
The results of the trial groups were compared by ANOVA analysis, and differences were considered significant at P < 0.05. Fisher's exact test was used to compare survival rates of mice in experimental and control groups.
Virus and mice
Plasmid construction
DNA transfection and immunoblotting analysis
Immunization and virus challenge
Detection of MCMV-specific neutralizing antibody
Immunofluorescence assay
Titration of MCMV in spleen
Interferon-γ ELISPOT assay
Statistical analysis
-
To test the efficacy of the gM/gN DNA vaccines, BALB/c mice were divided into seven groups and vaccinated with vector pDNA, pgB, pgM, pgN, or a combination of pgM and pgN at a dose of 50 μg for each DNA. Three weeks after the last immunization, the mice were challenged with a lethal dose of SG-MCMV (5×LD50). On day 5 post-challenge, three mice from each group were euthanased, and the spleens removed aseptically for measurement of viral titer. The remaining 10 mice from each group were monitored for 21 days for weight loss and survival.
As shown in Table 1, all mice in the negative control group G (3×pV50) died within the first week after challenge, mice in the positive control group A (3×pgB50) had a survival rate of 50%, and mice immunized with pgM (group B: 3×pgM50) or pgN (group C: 3×pgN50) alone had a survival rate of 30% or 20%, respectively. For the co-immunization groups, mice immunized three times with the high dose (group D: 3×pgM/pgN50) were fully protected, and their survival rate was 100%, whereas reducing the number of co-immunizations by one-third (group E: 2×pgM/pgN50) or the dose by one-half (group F: 3×pgM/pgN25) resulted in the survival rate dropping to 70% and 30%, respectively. In addition, mice in the negative control group showed clear signs of infection after challenge, presenting as lethargy, piloerection, anorexia, and emaciation. The first of the negative control mice died on day 3 and all died within 1 week after viral challenge, whereas the mice in the immunized groups survived for longer than those in the negative control group (Fig. 1A).

Table 1. Protection provided by gM, gN, or gM + gN DNA vaccines against lethal SG-MCMV challenge in mice after immunizationa
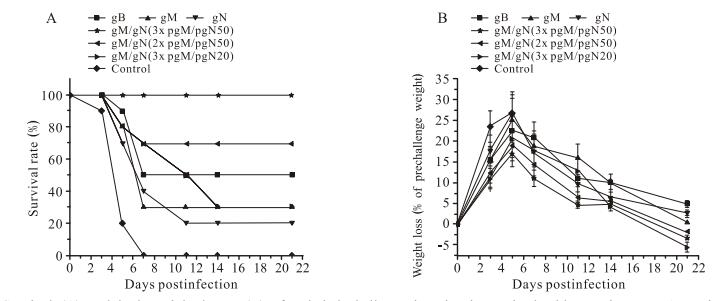
Figure 1. Survival (A) and body-weight losses (B) after lethal challenge in mice immunized with pgM/pgN DNAs. Mice were immunized with pgB, pgM, or pgN, respectively, or co-immunized with gM/gN pDNAs at a dose of 50 μg. Three weeks later, mice were infected with a lethal dose of SG-MCMV. Survival and body-weight changes of the mice were monitored 21 days post-challenge. Data points represent mean ± SD of each group in(B).
When the residual virus load in the spleen on day 5 post-challenge was compared, the titers of groups A, B, D, E, and F were significantly lower than that in the negative control group G, while the titer of group C (3×pgN50) was not significantly different from that of the control group. Further, when the immunized groups were compared, the pgM/pgN co-immunization groups had lower viral titers than the pgM or pgN group, and viral load decreased as the immunization dose increased. In particular, viral titer in group D (3×pgM/pgN50) was about 100 times lower than that in the negative control group G (Table 1), indicating that the immune responses induced by pgM/pgN coimmunization were able to effectively clear the virus in mice.
After challenge, the body weight of the mice changed in parallel with the change in other health indicators. Mice in the negative control group had the highest weight loss, nearly 30%. Mice in the single-antigen immunization groups showed clear symptoms of infection after challenge, and their weight loss was obvious but slightly less than the negative control group. Mice in the pgM/pgN co-immunization groups had milder symptoms of infection and less weight loss than mice in the single-antigen immunization groups, indicating that co-immunization provided mice with better protection (Fig. 1B).
-
Two weeks after the last vaccination, serum samples were collected for determination of the NAb levels elicited by pgM/pgN immunization. As shown in Fig. 2, no NAb could be detected in sera from the negative control group. For the single-antigen groups, the relative NAb titer levels in immune sera were: pgN < pgM < pgB. NAb titer levels in pgM/pgN groups were higher than that in the pgM or pgN group, and increased in line with the immunization times and the doses. NAb titers in groups D (3×pgM/pgN50) and E (2×pgM/pgN50) were significantly higher than that in the pgM or pgN groups, indicating that co-immunization of pgM and pgN induced stronger NAb response than immunization with each antigen alone.
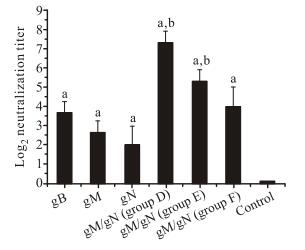
Figure 2. Neutralization titers of sera obtained from pgM/pgN immunized mice. Mice were vaccinated with gB, gM, or gN alone, or a mixture of gM/gN pDNAs at a dose of 50 μg. Sera were collected 2 weeks later and tested. Results were the highest serum dilutions at which 50% reduction in viral titer was achieved. Values represent the means ± SD of each group. aSignificant difference (p < 0.05) versus the mice in the negative control group. bSignificant difference (p < 0.05) versus the mice in gM or gN groups.
Immunoblot assay was performed to determine the specificity of the gM or gN immune sera. 293T cells were transfected with pgM or pgN alone or in combination (pgM/pgN). Cell lysates from MCMV-infected 3T3 cells were used as positive control. The polyclonal antiserum against gM reacted with proteins of approximately 86 kDa and 32 kDa in 293T cells transfected with pgM or pgM/pgN (Fig. 3A, lanes 2 and 3), and the lysates from MCMV-infected 3T3 cells also showed bands of a similar molecular weight (Fig. 3A, lane 5). Anti-gM sera also recognized a band with a molecular weight of about 125 kDa in transfected cells, along with the 86 kDa band mentioned above, which may represent the aggregates of gM protein, as gM is highly hydrophobic and insoluble, and readily forms high-molecular-weight aggregates (Fig. 3A, lanes 2 and 3). Fig. 3B showed that anti-gN sera identified a band of around 48 kDa from 293T cells transfected with pgN or pgM/pgN (Fig. 3B, lanes 2 and 3), similar to the band in infected 3T3 cells (Fig. 3B, lane 5). Anti-gN sera also reacted with a protein of around 15 kDa in transfected cells, which may be the gN form without post-translational modification (Fig. 3B, lanes 2 and 3). This was consistent with the previous finding that HCMV gN is a highly glycosylated envelope protein which is translated into an 18-kDa protein before being posttranslationally processed (Mach M, et al, 2000). When anti-gM/gN polyclonal sera were used, the 48 and 32 kDa bands were detected simultaneously in pgM/pgN-cotransfected 293T or MCMV-infected 3T3 cells (Fig. 3C, lanes 1 and 3). According to previous studies, HCMV gM and gN are able to form a complex with a molecular weight of 70 to 100 kDa (Mach M, et al, 2000). In the current study, lysates from MCMV pgM/pgN co-transfected 293T cells also showed some bands of between 70 and 100 kDa in size, similar to the bands in MCMV-infected 3T3 cells. The western blot results suggested that both the gM and gN DNA vaccines were able to express the proteins in mammalian cells, and that the anti-sera from gM or gN pDNA immunized mice had fine specificity. These anti-sera were also tested in immunofluorescence assays in MCMV-infected cells. As shown in Fig. 4, MCMV-infected 3T3 cells showed clear morphological changes, with the cell shape changing from the typical fibroblast shape to expanded round cells, the nucleus changing from oval to irregular, and some adjacent cells congregating and forming syncytia. Detection was first performed using anti-sera from pgM or pgN immunized mice, respectively, and this showed that in infected cells, gM or gN proteins were mainly distributed in the juxtanuclear regions rather than throughout the entire cytoplasm. A similar result was obtained with anti-sera from the pgM/pgN co-immunization group; the gM/gN complex proteins were mainly distributed in the juxtanuclear regions and not in the nucleus. The immunofluorescence results also indicated that the gM/gN immune sera had good specificity.
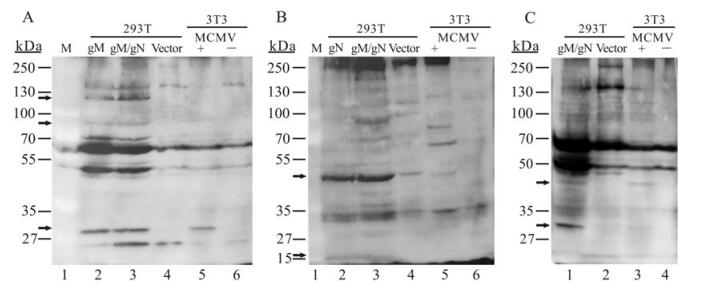
Figure 3. Immunoblotting analysis of transiently expressed gM and gN proteins with antisera to gM and gN. 293T: cell lysates of 293T cells transfected either with the vector DNA, pgM, or pgN, or co-transfected with pgM/pgN. 3T3: cell lysates of 3T3 cells infected (+) or not infected (-) with MCMV Smith strain. Lysate proteins were resolved on 10% SDS-PAGE gels, transferred to PVDF membranes, and immunoblotted with (A) polyclonal anti-gM, (B) anti-gN or (C) anti-gM/gN sera.

Figure 4. Confocal microscope analyses of 3T3 cells infected with MCMV Smith strain. Infected 3T3 cells were harvested 3 days post-infection and then fixed, permeabilized, and stained for immunofluorescence. Anti-gM/gN was detected with FITCconjugated anti-mouse IgG. Nuclei were stained with Hoechst 33258. Specific immunofluorescence was observed with a confocal laser scanning microscope. (Scale bar =10 μm).
-
To further define whether immunization with pgM/pgN can induce cellular immune responses, an ELISPOT assay was conducted. Cell-mediated immunity (CMI) to DNA vaccines was measured by quantifying the number of IFN-γ secreting splenocytes in immunized mice. Mice were inoculated three times with pgM, pgN, or pgM/pgN at a dose of 50 μg each. Data are presented as the average number of spots in triplicate stimulant wells.
The stimulant selected was inactivated MCMV virions, as gM is the most abundant virus glycoprotein and is present on the virus envelope in the form of gM-gN complex. As shown in Fig. 5, immunization with the pgM or pgN vaccine was able to induce some level of cellular immune response, and pgM induced a stronger response than pgN. For mice co-immunized with pgM/pgN, the number of specific IFN-γ-secreting splenocytes was significantly higher than that in mice immunized with pgM or pgN alone, being nearly four times higher than in pgM-immunized mice, thus indicating that pgM/pgN co-immunization was able to induce stronger cellular immune responses. In the negative control group, stimulation of splenocytes with inactivated MCMV resulted in only a few non-specific spots (spots ≤10 per 106 cells), which was similar to the background value of the ELISPOT plates (spleen cells without antigen stimulation). When splenocytes were stimulated with concanavalin, the non-specific IFN-γ positive spots were up to 2000 per 106 cells (data not shown).
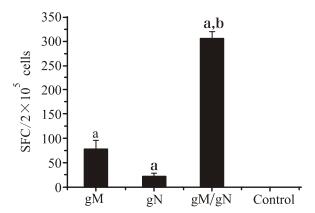
Figure 5. Cellular immune responses of mice vaccinated with gM/gN DNA vaccines. Mice were immunized with gM or gN or a mixture of gM and gN pDNAs at a dose of 50 μg. The control group was inoculated with vector plasmid. Splenocytes were isolated 2 weeks later and stimulated in vitro with 10 μg/mL of the inactivated MCMV. The results represent the geometrical means ± SD of each group. a Significant difference (p < 0.05) versus the mice in control group. b Significant difference (p < 0.05) versus the mice in gM or gN groups.
Protection provided by pgM-pgN immunization against lethal MCMV infection
Neutralizing antibody response
Cell-mediated immunity
-
The gM protein is highly conserved within the Herpesviridae family, and is a typical type Ⅲ membrane protein with six to eight hydrophobic transmembrane domains (Mach M, et al., 2000; Scalzo AA, et al., 1995). The gN protein is also conserved among the Herpesviridae family and contains multiple glycosylation sites. gN and gM can form a complex via covalent disulfide binds. Deletion of gM and/or gN has little effect on the in vitro replication of Alphaherpesvirinae, but insertional inactivation of gM gene results in replication deficiency in HCMV (Hobom U, et al, 2000; Jons A, et al., 1998). It has been reported that most domains of gM are required for gM-gN complex formation, and the disulfide linkage lies between the cysteine at position 44 of gM and cysteine 90 in gN (Mach M, et al., 2005). Sera from HCMV-seropositive populations were found in tests to be mostly non-reactive to gM or gN protein expressed separately, but 62% of samples were reactive to the gM-gN complex (Mach M, et al, 2000; Shimamura M, et al., 2006), indicating that this complex is an important target for the anti-viral humoral immune response. As both gM and gN are highly conserved proteins and gM is the most abundant viral glycoprotein, the gM-gN complex is an attractive candidate vaccine antigen. We studied the immunogenicity and protective effect of gCII complex antigens against MCMV challenge in a mouse model.
To our knowledge, the current study is the first report of testing of the pgM/pgN vaccine effect in an in vivo challenge model. We found that co-immunization with pgM and pgN three times at a high dose was able to provide mice with full protection against a lethal SG-MCMV challenge. Meanwhile, only 30% or 20% of the mice immunized with the gM or gN DNA vaccine alone survived viral challenge. The protection obtained with the gM-gN co-immunization was far greater than each antigen alone, which shows clear synergy between them. Studies have demonstrated that complex formation by gM and gN is necessary for the two proteins to complete post-translational modification and intracellular translocation (Mach M, et al, 2000; Mach M, et al, 2005), which might be an important reason for the excellent synergy seen with the gM/gN co-immunization in the current study. Shen et al. also found that co-immunization of gM and gN is necessary for inducing a good HCMV-specific NAb response (Shen S, et al, 2007).
Induction of NAbs is an important objective of CMV vaccine development, and envelope glycoproteins are key target antigens for inducing NAbs. In our experiments, the gM or gN DNA vaccines alone were able to induce NAbs, but at a low titer, whereas the gM/gN pDNA co-immunization induced a higher NAb titer than either component alone, and the titer increased in line with the immunization dose. The increase in NAb titer was also consistent with the decrease in residual virus load after challenge. In addition, the specificity of the gM/Gn immune sera was confirmed by immunoblotting and immunofluorescence assays. The western blot results showed that both the gM and gN DNA vaccines were able to express the proteins in mammalian cells, and the expressed proteins were similar to the viral proteins seen in MCMV-infected 3T3 cells. The gM and gN-homologous proteins are highly conserved in the Herpesvirus family, and they form a complex through disulfide bonds. HCMV gM and gN are able to form a complex with a molecular mass of 70 to 100 kDa in 293T cells co-transfected with pgM/pgN (Mach M, et al, 2000). In the current study, lysates from 293T cells co-transfected with MCMV pgM/pgN also showed some bands that migrated between 70 and 100 kDa, and similar bands were also detected in virus-infected cells. However, owing to the lack of mAb against the MCMV gM or gN protein, we cannot be certain that the bands were the gCII complex. Immunofluorescence assays showed that in MCMV-infected cells the gM and gN proteins shared the same cellular distribution at juxtanuclear regions and were not present in the nucleus. We therefore infer that in infected cells, gM and gN proteins are mainly localized in the cytoplasmic virus assembly compartment (AC) unique to Betaherpesvirusinfected cells, where AC are mainly distributed in the perinuclear region (Tandon R, et al, 2012). Viral tegument proteins and envelope constituents, including gB, gM/gN and gH/gL/gO, all assemble in the AC, where they fulfill the final processes in virus maturation and envelopment.
An important finding of this study is that coimmunization with pgM/pgN was able to induce cellular immune responses effectively, which has not been reported previously. Cellular immune responses play an important role in clearing CMV infection in vivo. In our experiments, immunization with pgM or pgN alone induced a moderate cellular immune response while pgM induced a stronger response than pgN, and the pgM plus pgN co-immunization induced significantly higher responses than pgM or pgN alone, indicating that the gM-gN complex vaccine is capable of inducing stronger cellular immune responses. Regarding the synergic effect of pgM/pgN co-immunization in induction of immune responses, this may be due to more B and T cell epitopes being provided by two proteins than by a single protein. Alternatively, the gCII complex formed by the gM and gN proteins might produce some new conformational epitopes, a notion that is supported by studies showing that some CMV antibodies were specific for protein complex epitopes but not for epitopes of the component proteins (Loomis, RJ, et al, 2012), and that epitopes with more diversity could induce better humoral and cellular immune responses, thereby providing better immune protection.
Clinical evidence has demonstrated that lowering virus load offers significant therapeutic benefits for patients with diseases caused by HCMV infection (Khanna, R, et al., 2006). An HCMV vaccine is the most practical approach to achieve this goal of lowering virus load. In the current study, the viral titers in the spleens of immunized mice were lower than those in the spleens of negative control mice. For mice co-immunized three times with pgM plus pgN, viral titer was nearly 100 times lower than that in the control group, indicating that the protective immune responses induced by gC Ⅱ complex DNA vaccine could effectively clear the virus infection in mice.
The gB has a certain degree of polymorphism in different HCMV clinical isolates, whereas the sequences of gM and gN are highly conserved (Shen S et al, 2007), but whether the DNA vaccine expressing gM and gN of MCMV Smith strain might cross-protect against other MCMV strains awaits to be validated in further experiments. However it is likely that inclusion of conserved protective antigens such as gM and gN into a CMV vaccine would provide broader protection against infection by different CMV strains.
In summary, the present study demonstrated by in vivo immunization and challenge experiments that the gC Ⅱ complex (gM/gN) DNA vaccine could effectively induce neutralizing antibodies and cellular immune response, and immunizing three times with a higher dose could provide mice with complete protection against a lethal MCMV challenge. The CMV gC Ⅱ complex is therefore a feasible candidate antigen for vaccines against CMV infection.
-
This study was supported by the Innovation Platform Open Fund of Hunan Provincial Education Department (11K010) and a research fund from Hunan Provincial Science and Technology Development (2008TP4033-2).


 DownLoad:
DownLoad: