









-
Hepatitis B virus (HBV) DNA synthesis occurs by reverse transcription of viral 3.5kb pregenomic RNA (pgRNA) synthesized from the nucleocapsid promoter (Nassal M, 2008; Ondracek C R, et al., 2011; Ondracek C R, et al., 2009; Ondracek C R, et al., 2009). Therefore, it is apparent that controlling the pgRNA level is a key regulatory step both in viral gene expression and in viral replication (Li L, et al., 2009; Tang H, et al., 2001). Liverenriched nuclear receptors (NRs) have been demonstrated to be essential for HBV RNA synthesis (Ondracek C R, et al., 2009; Ondracek C R, et al., 2009; Reese V C, et al., 2013; Shlomai A, et al., 2008). Complementation studies utilizing the human embryonic kidney cell line 293T, which support neither viral 3.5kb pgRNA synthesis nor HBV replication, revealed that only the NR hepatocyte nuclear factor 4α (HNF4α) and RXRα/PPARα (retinoid X receptor α plus peroxisome proliferator-activated receptor α) are capable of rescuing viral biosynthesis (Reese V, et al., 2011; Tang H, et al., 2001). These observations indicate that nuclear receptors may have a unique capacity to regulate HBV transcription and replication during natural infection. Indeed, it has been reported that HBV biosynthesis is likely to be completely dependent on HNF4α in vivo using a transgenic mouse model of chronic HBV infection (Ondracek C R, et al., 2011; Reese V, et al., 2011).
HNF4α and its coactivator are important regulators of energy homeostasis within the liver (Rodgers J T, et al., 2005; Yoon J C, et al., 2001). Peroxisome proliferatoractivated receptor-γ coactivator 1α (PGC1α), specifically and robustly coactivates key gluconeogenesis genes (Yoon J C, et al., 2001). Similarly, the amounts of HBV transcripts driven by the nucleocapsid promoter have also been shown to increase due to the coactivation effect of PGC1α and HNF4α (Shlomai A, et al., 2006). Tandem mass spectrometry analysis has shown that PGC1α is acetylated at 13 lysine sites. Mutation of these 13 lysines to arginines abolished the acetylation of PGC1α. If only some of the sites were mutated, PGC1α could still be still acetylated (Rodgers J T, et al., 2005). General Control Non-repressed Protein 5 (GCN5) acetylates PGC1α, resulting in a transcriptionally inactive state that relocalizes from promoter regions to nuclear foci. This process inhibits gluconeogenesis and is particularly important for the maintenance of glucose homeostasis. Adenovirus-mediated expression of GCN5 in cultured hepatocytes and in mouse liver significantly represses activation of gluconeogenic enzymes and decreases hepatic glucose levels (Doitsh G, et al., 2004; Lerin C, et al., 2006). However, there is still no direct study on the effect of GCN5 on HBV transcription and replication enhanced by PGC1α and HNF4α.
On the basis of these considerations, it was of interest to determine whether the acetyltransferase GCN5 might modulate HBV pgRNA synthesis and viral replication by regulating PGC1α activity. To address this issue, the expression vectors bearing PGC1α, acetylation site mutant PGC1αR13, GCN5 and acetyltransferase inactive mutant GCN5m were transferred into human hepatoma cell line Huh-7 and BALB/C mouse to evaluate the effects in vitro and in vivo respectively. Our results clearly show that GCN5, depending on the acetyltransferase activity, inhibits the coactivation activity of PGC1α in HBV transcription and replication.
HTML
-
The 1.3× wtHBV plasmid (1.3× wtHBV) was constructed by inserting 1.3 copies of the HBV genome (adw strain) with Eco RV site (5' terminus) and Taq Ⅰ site (3' terminus) into pGEM-3Z plasmid between the Sma Ⅰ and Acc Ⅰ sites (Doitsh G, et al., 2004). The 1.3× HBV-luc construct (HBV-luc) was derived from 1.3× wtHBV by substituting HBV sequences between the Bgl Ⅱ and Spe Ⅰ sites with the luciferase ORF (Doitsh G, et al., 2004). The pcDNA-Flag-PGC1α, pcDNA-Flag-PGC1αR13, pcDNA-Flag-GCN5, pcDNA-Flag-GCN5m and pcDNA-Flag-HNF4α plasmids express mouse PGC1α, GCN5, HNF4α or their corresponding mutant cDNA respectively with FLAG-tag, using the cytomegalovirus immediate-early promoter (Lerin C, et al., 2006; Rodgers J T, et al., 2005). The PGC1αR13 is mutated PGC1α with 13 potential acetylation sites changed from lysine to arginine (Rodgers J T, et al., 2005). GCN5m is an acetyltransferase catalytically inactive mutant of GCN5 (Liu X, et al., 2003). These constructs are individually labeled as 1.3× wtHBV, HBV-luc, p-GCN5, p-GCN5m, p-PGC1α, p-PGC1αR13 and p-HNF4α. A-1227/+57 G6Pase-luciferase reporter plasmid (G6Pase-luc) was constructed as described previously by Dieter Schmoll (Schmoll D, et al., 1999). pSUPER vector was used to construct siRNA plasmid against human GCN5 (GCN5 siRNA) and against mouse PGC1α (PGC1α siRNA). Due to the high homology of PGC1α sequences between human and mouse, the mouse PGC1α is also able to target human PGC1α (Li X, et al., 2007; Shlomai A, et al., 2006). The applied siRNA sequences were GCN5 siRNA 5'-tgttcgagctctcaaagat -3' and PGC1α siRNA 5'-ggtggattgaagtggtgta-3' (Lerin C, et al., 2006; Terreni M, et al., 2010).
-
The human hepatoma cell line Huh-7 was maintained in Dulbecco's modified Eagle's minimal essential medium as described previously by Ling Qiao (Qiao L, et al., 2013). Transfection was performed in 10 cm plates containing about 1×106 Huh-7 cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instruction. The following constructs were applied in transfection with the indicated combinations, 1.3× wtHBV (3 μg), HBV-luc (3 μg), G6Pase-luc (3 μg), p-HNF4α (3 μg), p-PGC1α (6 μg), p-PGC1αR13 (6 μg), p-GCN5 (9 μg), p-GCN5m (9 μg), GCN5 siRNA (9 μg) and PGC1α siRNA (9 μg). The same set of vectors lacking the corresponding target gene inserts were used in transfection as controls. When appropriate, a final concentration of 50 mmol/L nicotinamide (Sigma) was used to treat the cells for acetylation.
-
Female BALB/C mice (6 to 8 weeks of age) were kept under standard pathogen-free conditions in the Central Animal Laboratory of Wuhan Institute of Virology, Chinese Academy of Sciences, and treated under the institutional animal ethical standards. Mice were injected on day 0 with 1.5 mL of saline (0.9% NaCl) containing the indicated constructs or corresponding controls by hydrodynamic injection. The following plasmids were applied, 1.3× wtHBV (1 μg), p-PGC1α (2 μg) and p-GCN5 (3 μg). After 48h, animals were euthanized, and RNA and DNA were isolated from liver tissues for Northern and Southern blot analyses.
-
Huh-7 cells were transfected with the following constructs, HBV-luc, G6Pase-luc, p-HNF4α, p-PGC1α, p-PGC1αR13, p-GCN5 or p-GCN5m, where appropriate. Luciferase reporter assay was performed at 48h post-transfection using the luciferase assay system (Promega) according to the manufacturer's instruction. Results are shown as the fold increase of luciferase activity normalized to Renilla luciferase activity to adjust for transfection efficiency.
-
For Southern blot analysis, total liver DNA or core particle DNA from cells were prepared as described previously by Yongjun Tian (Tian Y, et al., 2011), and resolved by agarose gel (1.0% w/v) electrophoresis. For Northern blot analysis, transfected cells or liver tissues were homogenized in TRIzol (Invitrogen), and total RNA was isolated according to the manufacture's instruction. The isolated RNA was then mixed with formaldehyde-agarose gel loading buffer (Sigma) and heated at 65 ℃ for 5 min to denature the RNAs. The samples were then resolved in agarose gel (1.2% w/v) in the presence of formaldehyde, and electrophoresed for 6 hours at 50 V in formaldehyde-agarose gel running buffer (20 mmol/L MOPS, 5 mmol/L sodium acetate, 1 mmol/L EDTA, 20 mL 37% formaldehyde, RNase-free-water filled to 1000 mL, final pH 7.0). The resolved DNAs or RNAs were then blotted onto nylon membranes, and hybridized with 32P-labeled random-primed probe specific for HBV sequence. Bands were visualized by phosphorimaging using Cyclone Plus (Perkin Elmer). The cell amounts in Southern and Northern blots were respectively normalized by β-actin or 28S/18S amounts shown below the corresponding images.
-
For protein analysis, cells were lysed with 100 µL lysis buffer (50 mmol/L Tris-HCl pH 8.0, 150 mmol/L NaCl, 0.1% SDS, 1% NP40, 0.5% deoxycholic acid, 0.5% sodium azide and 100 µg/mL PMSF). The lysate of each sample was electrophoresed with 12% SDS-PAGE gel. After blotting onto polyvinylidene difluoride membranes (Immobilon P, Millipore), these were incubated with the indicated primary antibody and then the corresponding HRP conjugated secondary antibody. Bands were visualized using the DAB Western Blot kit (Thermo). The following antibodies were used, polyclonal rabbit anti-HBc (1:1000, DAKO), monoclonal anti-Flag (1:2000, Invitrogen), monoclonal anti-β-actin antibody (1:2000, Interchim), goat anti-mouse IgG and goat anti-rabbit IgG (1:3000, Proteintech, USA).
-
Statistical analyses were performed using unpaired two-tailed Student's t test. Differences were considered to be statistically significant when P < 0.05. All bar graphs are shown as the mean ± SE from at least 3 independent experiments and the images shown for Southern blot, Northern blot or western blot are the representative results from at least 3 independent experiments.
Plasmid constructions
Cells and transfections
Animal experiments
Luciferase assay
Southern and Northern blot
Western blot
Statistical analysis
-
There are 13 lysine sites in PGC1α which potentially can be acetylated (Rodgers J T, et al., 2005). In this study, we used a mutated construct p-PGC1αR13, which has mutations from lysine to arginine on all 13 potential acetylation sites. Because PGC1α was previously reported to coactivate HBV gene expression (Ardawi M S, et al., 1989; Shlomai A, et al., 2006), we wondered whether PGC1αR13 still has the coactivation activity with mutations at these sites. Toward this aim, Huh-7 cells were cotransfected with the 1.3× wtHBV construct, p-PGC1α or p-PGC1αR13. Northern blot analysis revealed that the amounts of all HBV transcripts were increased both in the presence of PGC1α, and PGC1αR13 also upregulated HBV transcription to a similar extent compared to PGC1α (Fig. 1A). Western blot analysis confirmed that the expression of viral core protein was also raised in the presence of PGC1α and PGC1αR13 (Fig. 1B). Next, the effect of PGC1α or PGC1αR13 on HBV replication was investigated. Southern blot analyzed the viral replicative intermediates from Huh-7 cells transfected with 1.3× wtHBV construct. The result clearly showed that PGC1αR13 and PGC1α also significantly upregulated HBV relaxed-circular (RC) and single-stranded (SS) DNA level (Fig. 1C). Furthermore, to quantify the impact of PGC1α or PGC1αR13 on HBV transcription, Huh-7 cells were transfected with the HBV-luc construct and analyzed for luciferase activity. The presence of the exogenous PGC1α or PGC1αR13 led to a significant increase in activity compared to the HBV-luc construct transfection alone (3.54±0.16 folds and 3.37±0.09 folds respectively). This induced increase was completely abolished by the application of siRNA against PGC1α, reducing luceferase activity to 0.61±0.02 folds. This also targeted PGC1αR13 (Fig. 1D). And the results indicate that PGC1αR13 also robustly coactivates HBV transcription as PGC1 in spite of the acetylation site mutations (Fig. 1D).
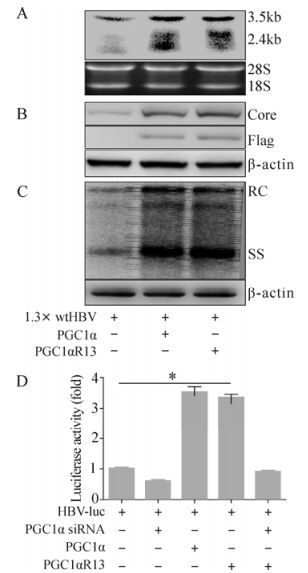
Figure 1. Enhancement of HBV transcription and replication by PGC1αR13. Huh-7 cells were transiently transfected with the indicated constructs together with 1.3× wtHBV and harvested for further analysis 72 hours post-transfection (A, B and C). A: Northern blot analysis (upper) for HBV transcripts with 18S/28S RNA amount normalization (lower). B: Western blot analysis (upper) for HBV core protein with β-actin normalization (lower). C: Southern blot analysis (upper) for HBV replicative intermediates with β-actin normalization (lower). D: Huh-7 cells were transfected with the indicated constructs together with HBV-luc and analyzed for luciferase activity 48 hours post-transfection. * p < 0.001.
-
PGC1α is known to be a strong transcriptional coactivator of the genes encoding PEPCK and G6Pase (glucose-6-phosphatase), the key gluconeogenic enzymes (Yoon J C, et al., 2001). So we subsequently analyzed the transcriptional coactivation activity of PGC1α on G6Pase with the luciferase reporter system. The G6Pase-luc construct with luciferase expression controlled by G6Pase promoter was used to assess the coactivation effect of PGC1α in the presence of HNF4α. After transfecting Huh-7 cells, HNF4α alone increased the luciferase activity up to 4.10±0.36 fold compared to control (Fig. 2A). PGC1α and PGC1αR13 further dramatically increased the luciferase activity up to 22.32±1.64 fold and 21.80±1.41 fold respectively in the presence of HNF4α, so the 13 sites mutation did not affect the coactivation activity (Fig. 2A). The nicotinamide (Nic), which acetylated PGC1α, inhibited the transcriptional activity of PGC1α/HNF4α and hence decreased luciferase activity from 22.74±1.72 fold to 1.26±012 fold, but not that of PGC1αR13 (18.10±0.95 fold) (Fig. 2B) due to the acetylation site mutations. As demonstrated previously, overexpression of the specific PGC1α acetyltransferase GCN5 increased the endogenous PGC1α acetylation level and further blocked the induction ability of PGC1α on gluconeogenic gene expression (Rodgers J T, et al., 2005). This has been confirmed by our data. GCN5 strongly repressed PGC1α transcriptional coactivation and reduced luciferase activity from 22.62±1.53 fold to 3.86±0.63 fold, but not for PGC1αR13 (19.70±1.30 fold) (Fig. 2C). It has been shown that GCN5 siRNA reduced intracellular GCN5 protein levels and further decreased PGC1α acetylation, which indicated the requirement of endogenous GCN5 for PGC1α acetylation (Lerin C, et al., 2006). Consistent with this observation and our previous result that GCN5 overexpression inhibited PGC1α transcriptional coactivation, GCN5 siRNA increased luciferase activity by 1.58±0.09 fold (Fig. 2D). Notably, the GCN5 induced repression was dependent on its acetyltransferase activity, since the catalytically inactive mutant GCN5m failed to completely abolish PGC1α transcription coactivation (14.54±1.31 fold) (Fig. 2E). Taken together, these results suggest that GCN5 suppresses the coactivation activity of PGC1α in the process of HNF4α-associated G6Pase expression.
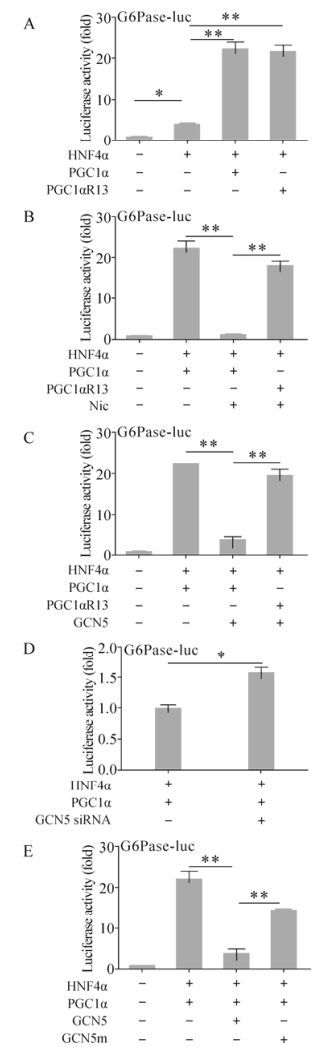
Figure 2. Suppression of PGC1α transcriptional coactivation activity through GCN5 acetyltransferase activity. Huh-7 cells were transiently transfected with the indicated construct combination together with G6Pase-luc. Where indicated, 50mM nicotinamide (Nic) was added to the cells 16 hours post-transfection. 48 hours post-transfection, cells were harvested and analyzed for luciferase activity. * p < 0.05; ** p < 0.001.
-
We have shown that GCN5 suppressed PGC1α coactivation activity on G6Pase expression in an acetyltransferase activity dependent manner. This prompted us to investigate whether GCN5 directly plays a role on PGC1α-related coactivation of HBV transcription and replication. Evaluation by HBV-luc showed similar results to G6Pase-luc. HNF4α alone increased luciferase activity in the HBV-luc transfected Huh-7 cells 2.29±0.33 folds as expected, and the exogenous PGC1αR13 further enhanced it up to 5.53±0.17 fold, similar to PGC1α (5.92±0.39 fold) (Fig. 3A). GCN5 dramatically repressed the PGC1α-induced luciferase activity by 2.08±0.21 fold, whereas GCN5m didn't have such an effect (4.68±0.23 fold) (Fig. 3B). In contrast, PGC1αR13 didn't respond to GCN5 (5.55±0.17 fold) (Fig. 3C) because of the acetylation site mutations. Knocking-down endogenous GCN5 by siRNA increased PGC1α/HNF4α enhanced HBV-luc activity by 5.87±0.28 fold to 7.54±0.37 fold (Fig. 3D). Analysis of HBV transcripts and replicative intermediates within 1.3× wtHBV construct transfected Huh-7 cells confirmed the HBV-luc results. Northern blot and Southern blot analysis clearly showed that HBV transcription and replication were enhanced by PGC1α/HNF4α, and GCN5 reduced the enhancement (Fig. 3E and F). In contrast, due to the loss of acetylation activity, GCN5m had no influence on PGC1α-related HBV replication enhancement (Fig. 3F). These results indicate that GCN5 represses the enhancement effect of PGC1α/HNF4α on HBV transcription and replication by acetylating PGC1α.
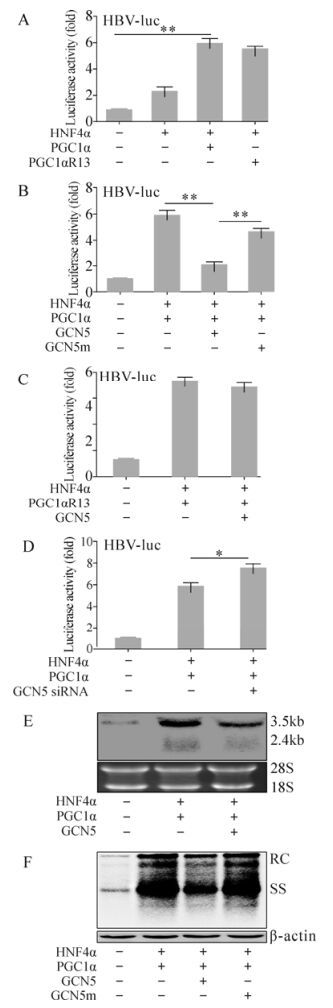
Figure 3. GCN5-mediated suppression of PGC1α-induced HBV biosynthesis enhancement in vitro. (A, B, C and D) Huh-7 cells were transfected with the indicated constructs together with HBV-luc and analyzed for luciferase activity 48 hours post transfection. * p < 0.05; ** p < 0.001. (E and F) Huh-7 cells were transfected with the indicated constructs together with 1.3× wtHBV and analyzed by Northern blot (E, upper) and Southern blot (F, upper) 72 hours post transfection. 18S/28S RNA (E, lower) and β-actin (F, lower) were used for cell amount normalization.
-
Finally, to confirm the potential GCN5/PGC1α epigenetic role during HBV biosynthesis in vivo, we treated BALB/C mice with 1.3× wtHBV, p-PGC1α and p-GCN5 via hydrodynamic injection into the tail vein and kept them under normal feeding conditions. Total RNA and DNA were isolated from mouse liver and analyzed by Northern and Southern blot at 48 hours post injection. Consistent with the aforementioned in vitro results, HBV transcripts (Fig. 4A) and replicative intermediate DNA (Fig. 4B) were clearly increased in the mice treated with exogenous PGC1α, which were partially down-regulated in the presence of GCN5, in spite of weaker repression than that observed in Huh-7 cells. In summary, these results proved that GCN5 also represses PGC1α-induced enhancement of HBV transcription and replication in vivo.
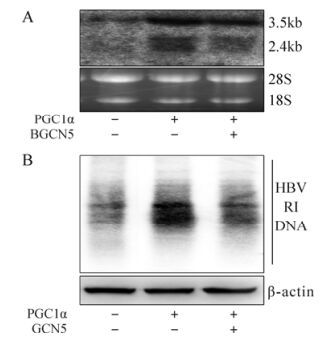
Figure 4. GCN5-mediated suppression of PGC1α-enhanced HBV transcription and replication in vivo. BALB/C Mice were injected the indicated constructs together with 1.3× wtHBV. Total RNA and DNA were extracted from the mice livers after 48 hours. A: Northern blot analysis (upper) for HBV transcripts with 18S/28S RNA normalization (lower). B: Southern blot analysis (upper) for HBV DNA replicative intermediates (RI) with β-actin normalization (lower).
PGC1αR13 coactivates HBV gene expression
GCN5 represses PGC1α transcriptional coactivation activity
GCN5 inhibits PGC1α-induced HBV transcription and replication in Huh-7 cells
GCN5 inhibits PGC1α-induced HBV biosynthesis in vivo
-
HBV replicates by reverse transcription of the viral 3.5 kb pgRNA (Nassal M, 2008). Hence, by controlling HBV transcription level it is possible to suppress both HBV replication and viral gene expression. Activation of nuclear receptor-mediated transcription by coactivators has been shown to play an important role in liver energy homeostasis (Reese V, et al., 2011; Yoon J C, et al., 2001). In particular, it has been demonstrated that the coactivator PGC1α is a critical determinant for both gluconeogenesis and HBV biosynthesis (Shlomai A, et al., 2006). It has been reported that the regulation of HBV replication and key metabolic functions may share the same signaling pathway in hepatocytes (Ondracek C R, et al., 2009; Shlomai A, et al., 2006). PGC1α phosphorylation by AKT/PKB (v-akt murine thymoma viral oncogene homolog/ protein kinase B) at serine 570 blocks its association with nuclear receptors, and further reduces the expression of G6Pase, PEPCK (phosphoenolpyruvate carboxykinase) and HBV pgRNA transcription (Li X, et al., 2007; Ondracek C R, et al., 2011). By purifying endogenous PGC1α complexes, GCN5 has been identified as an essential component in the PGC1α transcriptional pathway, which negatively regulates the expression of gluconeogenic genes by directly acetylating PGC1α (Lerin C, et al., 2006). These observations have raised the hypothesis that HBV transcription and replication might be similarly regulated. However, to date, only the effects of PGC1α and major phosphorylation mutant PGC1αS570A on HBV transcription and replication have been examined in detail (Guo H, et al., 2007; Ondracek C R, et al., 2011).
In the current study, the effect of GCN5 on PGC1α transcriptional activity was evaluated using G6Pase-luc, HBV-luc, as well as 1.3× wtHBV constructs both in vitro and in vivo. Overexpression of PGC1α specific acetyltransferase GCN5 inhibited the transcriptional activity of PGC1α/HNF4α (Fig. 2C, Fig. 3B, E and F), while decreasing endogenous GCN5 by GCN5 siRNA enhanced its transcriptional activity (Fig. 2D, Fig. 3D). GCN5 suppressed PGC1α/HNF4α transcriptional activity specifically depending on its acetyltransferase activity, since neither the acetyltransferase deactivated mutant GCN5m nor the acetylation site mutant PGC1αR13 have such an effect (Fig. 2E, Fig 3B, C and D). Notably, the in vivo analysis showed a similar phenomenon. PGC1α clearly increased HBV transcription and replication, and GCN5 partially reduced PGC1α-related enhancement (Fig. 4A and 4B). The suppression effect in vivo has not been seen as strongly as that observed in vitro, probably due to the complicated microenvironment in vivo. Generally, this study gives us a valuable clue to explore a novel mechanism for HBV-hepatocyte interaction involved in viral replication and major metabolic processes.
Overall, our evidence implies that the regulation of the GCN5/PGC1α pathway may play an essential role in modulating HBV biosynthesis under a variety of physiologically relevant conditions. Further understanding of GCN5-mediated regulation process during HBV transcription and replication may offer an insight into developing novel antiviral agents against potential targets in HBV infection. Furthermore, GCN5 may serve as a model regulator of modulating PGC1α-induced HBV transcription and replication through the acetylation of PGC1α. Finally, it will be of interest to investigate other signaling transduction pathways involved in the regulation of PGC1α activity, and their physiological roles in HBV transcription and replication in hepatocytes (Doitsh G, et al., 2004; Oropeza C E, et al., 2008; Potthoff M J, et al., 2009; Wang Y, et al., 2012).
-
We are grateful to Dr. Yosef Shaul for kindly providing the plasmids 1.3× wtHBV and 1.3× HBV-luc, and Dr. Pere Puigserver for the plasmids pcDNA-Flag-PGC1α, pcDNA-Flag-PGC1αR13, pcDNA-Flag-GCN5, pcDNA-Flag-GCN5m and pcDNA-Flag-HNF4α. This work was supported by grants from the National Major Science and Technology Special Projects for Infectious Diseases of China (2012ZX10004503-008, 2012ZX10001006-002, and 2012ZX10002006-002).


 DownLoad:
DownLoad: