









-
Coccolithoviruses are a group of viruses that infect Emiliania huxleyi, a coccolithophorid alga with a global distribution in temperate and sub-temperate oceanic regions, and therefore play a crucial role in biogeochemical cycling and primary productivity (van Rijssel M, et al., 2002). So far coccolithoviruses have been studied in mesocosm systems (Martinez J M, et al., 2007), natural open ocean blooms (Rowe J M, et al., 2011; Wilson W H, et al., 2002), and in laboratory based experiments (Allen M J, et al., 2006; Wilson W H, et al., 2005). With the recent sequencing of laboratory isolates, glimpses into the natural biodiversity of these viruses at the genetic level have been observed (Allen M J, et al., 2006; Nissimov J I, et al., 2011; Nissimov J I, et al., 2011; Nissimov J I, et al., 2012; Nissimov J I, et al., 2012; Pagarete A, et al., 2012; Wilson W H, et al., 2005). EhV-86, the model strain, harbours a 407, 339 bp genome which encodes 472 genes including core Nucleo-cytoplasmic large DNA Virus (NCLDV) genes for DNA polymerase, major capsid protein and RNA polymerase (Allen M J, et al., 2006). Despite the designation of the majority of the content as unknown or putative function, the genetic machinery for a near complete sphingolipid biosynthesis pathway (acquired through horizontal gene transfer from the host, E. huxleyi) has been identified in every coccolithovirus isolate to date (Allen M J, et al., 2006; Monier A, et al., 2009). Genomic analysis of coccolithoviruses isolated from different geographical locations has shown that, despite displaying similar genome sizes, they differ at a number of genomic loci; however the functional relevance of this has yet to be determined experimentally (Allen M J, et al., 2007). Problems have arisen when extrapolating the diversity characterised in the laboratory to naturally occurring environmental virus communities. Viral isolates studied in the laboratory often represent the most abundant virus strains present at the time of isolation (often dependant on the most abundant host at that time), and are biased towards isolates capable of infecting established laboratory strains of E. huxleyi and may or may not be environmentally relevant. Indeed, during natural conditions in the oceans, many different viral strains compete with each other for infection and replication. Some will be more successful than others. The success of a virus is determined by a plethora of transient environmental conditions which create an ever-changing landscape to adapt and evolve to. It is this intense selection pressure that contributes to the diverse pool of genes observed in the handful of 'model' strains characterised to date. However, reliance on a limited number of strains to infer ecological functional relevance often ignores the diversity and variation found in the natural environment.
Traditionally, the DNA polymerase gene is used to study the diversity and phylogeny of phycodnaviruses (algal viruses) (Chen F, et al., 1996). In recent years the gene encoding the major capsid protein (MCP) has also been used as an alternative marker, capable of distinguishing phylogenetic differences on a strain level (Larsen J B, et al., 2008; Rowe J M, et al., 2011). Several cruises have used these markers to observe the diversity of coccolithoviruses in natural blooms. The first looked at the temporal succession of E. huxleyi and their viruses during the propagation of a natural bloom in the North Sea in 1999 (Martinez J M, et al., 2012), whilst another cruise in the North Atlantic between Iceland and the UK in 2005 focused mainly on the distribution of coccolithoviruses, their location specific distinctions and their clustering with the use of the MCP marker gene (Rowe J M, et al., 2011).
Despite these efforts, there are many questions that still remain unanswered. For example, it is not currently known how the coccolithoviruses persist during non-bloom periods. With the exception of coccolithovirus sequences extracted from Black Sea sediments (Coolen M J, 2011), the diversity of these viruses in non-bloom conditions is poorly understood. Given the harsh conditions to which viruses are exposed to in their natural environment, it is somewhat surprising that infection, and the resulting bloom termination, occurs regularly and reliably on a yearly basis. Yet, perhaps, the most important questions left unanswered (in this and the majority of virus systems under laboratory study) is what is the functional relevance of the observed biodiversity, and how does it impact on the ecology of the virus community and its function?
Here, we aim to investigate both the biogeographic and temporal distribution of coccolithoviruses and their diversity with the established MCP marker, whilst also targeting a gene whose protein is of known metabolic function during infection, serine palmitoyltransferase (SPT). SPT is the first and rate limiting enzyme in the de novo sphingolipid biosynthesis pathway and homologues are encoded by both the virus and host genomes (Han G, et al., 2006; Monier A, et al., 2009; Wilson W H, et al., 2005). It has been implicated in the formation of lipid rafts and virus release during infection (Pagarete A, et al., 2009), and is even considered to be involved in the mass termination of coccolithophore blooms via the propagation of programmed cell death (PCD) of its host (Bidle K D, et al., 2011; Vardi A, et al., 2009; Vardi A, et al., 2012). SPT gene expression has been observed during infection of E. huxleyi under both laboratory and natural conditions, and the enzyme's activity has been characterised (Allen M J, et al., 2006; Han G, et al., 2006; Pagarete A, et al., 2009; Pagarete A, et al., 2012). Here, we use the two genes as markers for phylogeny and functionality in a study attempting to assess both spatial and temporal variability, using an archive of DNA samples collected during a cruise in the Atlantic Ocean, a coccolithophore bloom cruise in the North Sea in 1999, and samples collected weekly during a seven year period from the Western Channel Observatory in the English Channel near Plymouth, UK. By obtaining samples from a variety of locations and time points, and using the two marker genes we were hoping to improve the current understanding of the classification of these viruses, their distribution, and gain an insight into their functional biodiversity and ecological relevance.
HTML
-
Seawater was collected twice a day (before dawn and at solar noon) through a Conductivity Temperature and Density instrument (CTD) at 65 stations along the Atlantic Meridional Transect-20 (AMT-20) cruise track (www.amt-uk.org) (Supplementary material Fig. S1 and S2). Samples (10 L) were collected from five depths at each station corresponding to 97%, 55%, 33%, 14% and 1% light penetration, filtered via a 0.2 μm Millipore nitrocellulose membrane filter (47 mm), snap-frozen in liquid nitrogen and stored at -80 ℃. Two 1 mL samples from each depth were fixed in 1% glutaraldehyde for Analytical Flow Cytometry (AFC) of coccolithophores and coccolithoviruses. AFC data are available from http://www.bodc.ac.uk/projects/uk/amt/.
-
The Western Channel Observatory is located 10 km south off Plymouth Sound in the English Channel (50° 15.00' N, 4° 13.02' W). Weekly samples (1 L) were taken between 2001 and 2007 from the L4 station, filtered onto 0.45 μm Millipore nitrocellulose membrane filter (47 mm), snap-frozen in liquid nitrogen and stored at -80 ℃. Two 1 mL samples were fixed in 1% glutaraldehyde for Analytical Flow Cytometry (AFC) of coccolithophores and coccolithoviruses.
-
Extracted DNA samples from the DISCO cruise (Dimethyl Sulphide Biogeochemistry within a Coccolithophore Bloom) were obtained from the Plymouth Marine Laboratory DNA archive. Samples were originally collected in June, 1999 on board the RRS Discovery during a phytoplankton bloom located in the North Sea (East to West from -2.0° to 4.0° and North to South from 61.0° to 51.0°). Further details on the methodology used can be obtained from Martinez et al (Martinez J M, et al., 2012).
-
All samples were subjected to a total genomic DNA extraction following an adapted phenol-chloroform protocol as described by Schroeder et al. (Schroeder D C, et al., 2005). Extracted DNA samples were subjected to a two-step nested PCR. Primers and reaction conditions for the detection of the coccolithovirus Major Capsid Protein (MCP) by DGGE have been described previously (Martinez J M, et al., 2012; Martinez J M, et al., 2007). Primers for the detection of the coccolithovirus serine palmytoyltransferase (SPT) gene were designed manually following the multiple DNA alignment of SPT from nine fully sequenced coccolithovirus genomes (EhV-84, EhV-86, EhV-88, EhV-99B1, EhV-201, EhV-202, EhV-203, EhV-207 and EhV-208). All PCR reactions were conducted in a VWR JENCONS Uno Thermal Cycler in 25 μL final volume (cycle conditions in Supplementary material Table S1). For the first step of the nested PCR reaction, 1 μL of DNA template (typically ~50 ng of extracted DNA) was mixed with 5 L of 5 × PCR reaction buffer (Promega), 1.5 L of 25 mmol/L MgCl2, 0.1 L of Taq DNA polymerase (Promega), 2 L of each 10 mol/L primer (MCP-F1/ MCP-R1 or SPT-F1/SPT-R1, see Table 1), 1.25 L of 2 mmol/L dNTPs and DNA-free molecular biology grade water (Sigma-Aldrich) up to a final volume of 25 L. Only samples that gave a band when visualised by agarose electrophoresis after the first step were subjected to the second PCR step. The second reaction of the nested PCR was performed under the same conditions as the first round, except 2 μL of product from the first reaction was used as template and primers used were MCP-F2-GC/MCP-R2 (generating a 135 bp fragment) or SPT-F2-GC/SPT-R2 (generating a 335 bp fragment), see Table 1.
Primer Sequence (5' to 3') MCP-F1 GTCTTCGTACCAGAAGCACTCGCT MCP-R1 ACGCCTCGGTGTACGC ACCCTCA MCP-F2-GC CGCCCGGGGCGCGCCCCGGGCGGGGCGGGGGCACGGGGGGTTCGCGCTCGAGTCGATC MCP-R2 GACCTTTAGGCCAGGGAG SPT-F1 GTTGGATATCCCGCAACACC SPT-R1 CAATGTCGCCAATGTTGGC SPT-F2-GC CGCCCGGGGCGCGCCCCGGGCGGGGCGGGGGCACGGGGGGGAATCTCGCGCGCG SPT-R2 CGCGGTCCACATGTACC SPT-F2 GGAATCTCGCGCGCG Table 1. The primers used in this study
DGGE was performed using an Ingeny PhorU-2 system. 15 μL of nested PCR product was applied directly onto an 8% w/v polyacrylamide gel (acrylamide /N, N'-methylene bisacrylamide, 37:1, w/w) in 1 × TAE buffer (40 mmol/L Tris pH 7.4, 20 mmol/L NaAcetate, 1 mmol/L Na2EDTA). A 30 to 60% linear denaturing gradient was formed using 20% and 80% denaturants (100% denaturant being 7 mol/L urea and 40% v/v formamide). 20 μL of marker (composed of the single product from the nested PCR of nine laboratory strains) was used in the first well of each gel. For SPT samples, electrophoresis was performed at a constant voltage of 100V and a temperature of 60 ℃ for 17 hrs. For MCP samples a constant voltage of 200V was used for 3.5 hrs. Following electrophoresis the gels were stained for 1 hr in Milli-Q water containing 1 μg/mL Ethidium Bromide then de-stained in Milli-Q water for 1 hr, visualised on a UV transilluminator (Syngene GeneGenius) and photographed using the Syngene GeneSnap software. Bands of interest were excised and incubated in 30 μL of DNA water at 4 ℃ overnight and then 2 μL was used as a template for a final third step PCR following all the conditions of the second step (using MCP-F2-GC /MCP-R2 or SPT-F2/SPT-R2, Table 1), prior to sequencing. Sanger sequencing was performed by the LGC Genomics Sequencing Centre in Germany (www.lgcgenomics.com). The LifeTech (former Applied Biosystems) BigDye version 3.1 sequencing mix was used for cycle sequencing. After the purification of the sequencing reactions by gel filtration (Centri–Pur 96, EMP biotech Berlin) the samples were run on an ABI 3730 XL instrument using POP7 polymer and standard run conditions.
-
Coccolithovirus isolate MCP and SPT sequences retrieved from GenBank (JF974290, AJ890364, JF974310, FN429076, JF974311, EhV-202, JF974291, JF974317 and JF974318) and the new environmental sequences (submitted to GenBank under accession numbers: AB738836, AB738837, AB738838, AB738839, AB738840, AB738841, AB738842, AB738843, AB738844, HE970437, HE970438, HE970439 and HE970440) were aligned using the MEGA4 (version 4.0.2) multiple sequence alignment software (Tamura K, et al., 2007). Prior to CLUSTALW alignment the primer sequences were removed and the sequences were made the same length in order to decrease bias that can arise from potential gaps and sequences with different lengths. The evolutionary history was inferred using the NeighborJoining method (Saitou N, et al., 1987). Phylogenetic analyses were conducted in MEGA4 (Tamura K, et al., 2007). Translated MCP and SPT gene sequences from EhV-86 and EhV-99B1 were modelled in order to determine their predicted secondary and tertiary protein structures. Sequences were uploaded into the Phyre2 protein fold recognition server (Kelley L A, et al., 2009). The final models of SPT and MCP were conducted by Phyre2 using seven and six templates respectively in order to maximise confidence, percentage identity and alignment coverage. The resulting theoretical models were uploaded into the 3D protein structure analysis software: Jmol (Hanson R, 2010), Swiss PdBViewer (Guex N, et al., 1997), and Astex (Hartshorn M J, 2002).
-
Host and virus abundance was determined using a FACScan Flow Cytometer (Beckton Dickinson, Oxford, UK) as described by Brussaard et al (Brussaard C P, et al., 2000). Data files were analysed using the WinMDI 2.9 and CellQuestProTM software.
Atlantic Meridional Transect Cruise (AMT) Sample Collection
The Western Channel Observatory (WCO) Time Series
DISCO Cruise Sample Collection (1999)
DNA Extraction, PCR and DGGE Analysis
Bioinformatic Analysis
Analytical Flow Cytometry of Coccolithophores and Coccolithoviruses
-
Samples were obtained from the Atlantic Meridional Transect (AMT) between Southampton, UK and Chile, over a 6 week period from 12th October to 25th November 2010 (AMT-20 cruise: www.amt-uk.org/cruises/amt20). In total, 325 samples were taken from 65 stations at 5 depths (Supplementary material Fig. S1 and S2). A further 117 surface seawater samples from the L4 station of the Western Channel Observatory (www.westernchannelobservatory.org.uk) were obtained between 2001 and 2007. The DImethyl Sulphide biogeochemistry within a COccolithophorid bloom cruise (DISCO) yielded an additional 144 DNA samples obtained in 1999 between 5th June and 1st July 1999 and are currently archived at PML. Analytical flow cytometry (AFC) analysis revealed virus like particles in most AMT and L4 samples (data not shown). However, no specific signature for coccolithoviruses was observed anywhere along the Atlantic transect, although coccolithophores were observed in low abundances (data not shown). DISCO samples, as intended, have previously been shown by Martinez et al to be dominated by coccolithophore and coccolithovirus signatures (Martinez J M, et al., 2012).
-
In total, 25% of the 325 (i.e. 80 samples) samples collected on the AMT-20 transect successfully amplified a coccolithovirus MCP product. Furthermore, the majority of positive samples displayed multiple distinct bands by DGGE analysis. MCP was detected at locations along the entire transect (Fig. 1), even in areas where host presence is not typically expected and only observed by AFC in small numbers (data not shown). The number of distinct bands observed by DGGE analysis decreased around the North and South Atlantic Gyres, as phytoplankton abundance also decreased. The highest numbers of distinct bands were consistently detected at the Deep Chlorophyll Maximum (DCM) region of each sampling station along the transect and there was a positive correlation between increased depth and number of distinct MCP bands observed (Pearson correlation, r=0.53) (Fig. 2). Of the 117 L4 samples, 103 (83%) produced a coccolithovirus MCP product. MCP was detected in all years, throughout the year. In the analysis by Martinez et al, the authors successfully amplified an MCP product from 132 of the 144 (i.e. 91%) samples from the DISCO series (Martinez J M, et al., 2012).
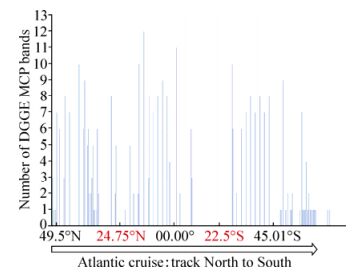
Figure 1. Number of MCP bands of samples on the AMT-20 transect, North to South, as detected by DGGE. Places in red indicate the centres of the North and South Atlantic gyres.
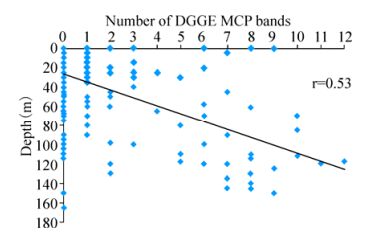
Figure 2. The number of MCP bands in each sample detected by DGGE on the AMT-20 transect cruise. There was a positive correlation of the number of distinct MCP bands with increasing depth (Pearson, r=0.53). 245 samples had no bands and 80 samples had one or more bands, increasing with depth.
Distinct bands were successfully extracted and sequenced from MCP DGGE gels. AMT samples yielded 33 distinct bands, and L4 samples a further 25 bands. Newly acquired sequences were used in combination with nine MCP gene fragments from characterised EhV isolates for phylogenetic analysis. Although DGGE analysis was based upon a 135 bp fragment, inconsistent sequencing quality in a minority of bands restricted subsequent analysis to a 50 bp subregion in order to allow the inclusion of all samples. Most AMT-20 and L4 sequences clustered into one of five sub-clades of coccolithoviruses: denoted as A, B, C, D or E, alongside established laboratory isolates (Fig. 3). However, nine new genotypes were discovered: AMT2b-3 [DDBJ: AB738836], AMT273-1 [DDBJ: AB738837], AMTt1-2 [DDBJ: AB738838], L4/23.04.2001/c [DDBJ: AB738839], L4/12.07.2006/a [DDBJ: AB738840], L4/04. 06.2007/a [DDBJ: AB738841], L4/02.01.2006 [DDBJ: AB738842], L4/02.09.2002 [DDBJ: AB738843], and L4/ 01.07.2003 [DDBJ: AB738844].
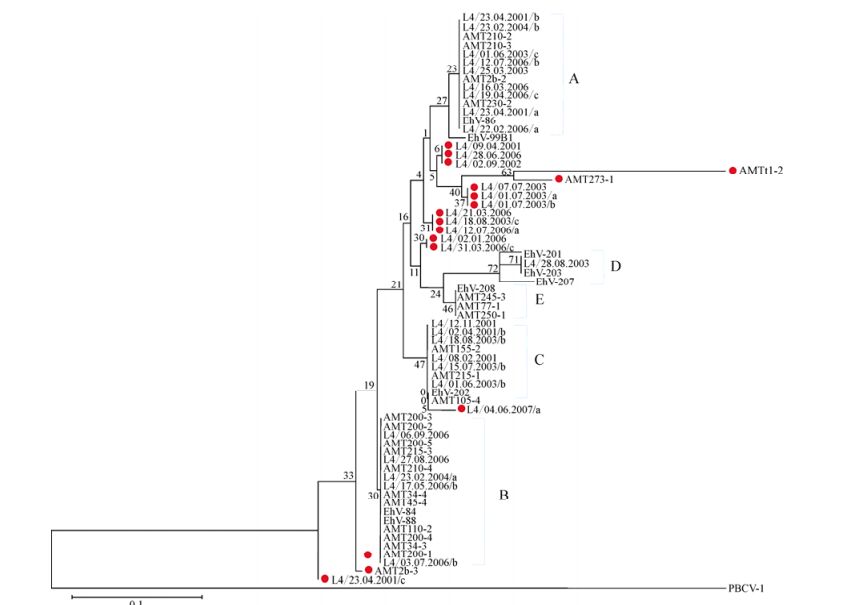
Figure 3. Evolutionary relationships of 69 MCP gene fragment (50 bases) sequences. The sequences were extracted from DGGE bands from the AMT-20 transect and the L4 time series and the nine sequences of laboratory EhV strains were obtained from NCBI database. Places in red indicate novel sequences that cluster outside of the five already known clades of EhV MCPs (A=EhV-86 like, B= EhV-84 and EhV-88 like, C= EhV-202 like, D=EhV-201, EhV-203 and EhV-207 like, and E=EhV-208 like). The optimal tree with the sum of branch length = 0.54677874 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (10, 000 replicates) is shown next to the branches. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site.
-
Despite the short MCP fragment the analysis was based upon, the coccolithoviruses displayed high diversity with nucleotide polymorphisms observed at 14 of the 50 nucleotide locations under analysis (corresponding to amino acids 54-69 of the MCP protein), which corresponded to just three amino acid changes when translated (Supplementary material Fig. S3 and S4). The majority of the novel environmental sequences differed from the EhV isolate sequences at one amino acid position; i.e. Phenylalanine replaced by Serine, or Valine replaced by Isoleucine. The AMT-20 sequence AMTt1-2 differed at two amino acids from the EhV sequences; Alanine replaced by Serine, and Valine replaced by Isoleucine. Using the full length MCP from EhV-99B1, we modelled the 3D protein structure of the predicted major capsid protein. 74% of the final model of the MCP protein was modelled with an accuracy of >90%, based on the following six protein structures on the Protein Databank in a decreasing order of alignment coverage and % of confidence: 1J5Q, 1M4X 1M3Ya1, 1M3Ta2, 3SAM, and 2W0C.
When modelled, the coccolithovirus diversity observed (within the domain equivalent to the Adenovirus Hexon subdomain 4) was predicted to have negligible, if any, impact on overall tertiary structure of MCP (data not shown). Indeed, even the potentially greatest change between the small, polar Serine and the large, hydrophobic Phenylalanine had no impact whatsoever on the predicted structure. This is most likely due to the high degree of structural conservation, regardless of the primary sequence, observed in all major capsid proteins and virions studied to date (Abrescia N G, et al., 2012; Bamford D H, et al., 2005; Krupovic M, et al., 2008; Krupovic M, et al., 2011).
-
Despite the success with the MCP marker, SPT gene fragments could not be amplified in any of the AMT-20 samples. However, of the 117 L4 samples, 41 (35%) successfully amplified a 335 bp product for the coccolithovirus SPT gene. Of the 144 DISCO cruise samples, 65 (45%) successfully amplified a product. 14 and 16 distinct bands were successfully extracted and sequenced from L4 and DISCO SPT DGGE gels, respectively. Phylogenetic analysis of a 207 bp region revealed that most L4 and DISCO sequences clustered into one of four sub-genotypes of the SPT gene: A, B, C, or D, and were similar to SPT sequences obtained from characterised EhV isolates (Fig. 4). However at least four new genotypes were discovered: DIS/29.06.1999/80m [EMBL: HE970437], DIS/23.06.1999/ 100m/a [EMBL: HE970438] (E); L4/16.09.2002 [EMBL: HE970439], and L4/15.07.2003 [EMBL: HE970440] (F).
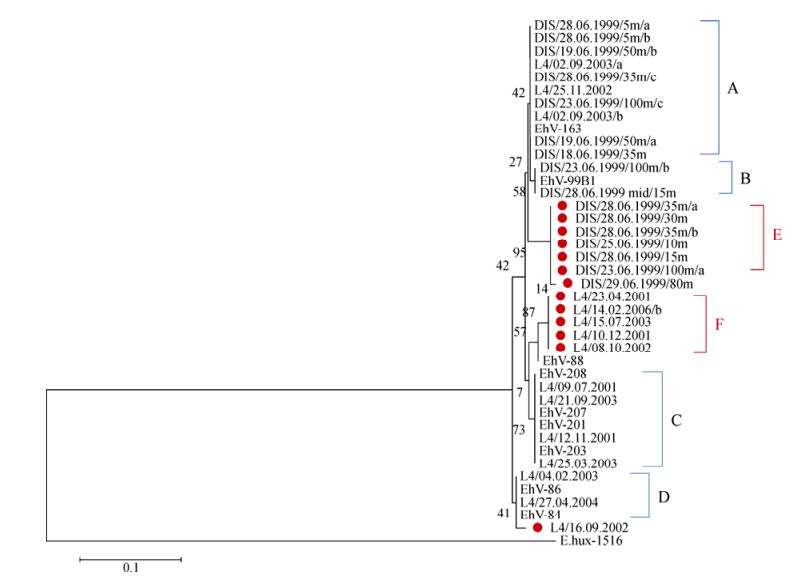
Figure 4. Evolutionary relationships of 41 SPT gene fragment (207 bases) sequences. The sequences were extracted from DGGE bands from the L4 time series and the DISCO cruise samples, and the nine sequences of laboratory EhV strains were obtained from the NCBI database. E. huxleyi CCMP 1516 SPT is used as an outlier. Places in red indicate novel sequences and that cluster outside of the four already known clades of EhV SPTs (A=EhV-163 like, B=EhV-99B1 like, C=EhV201, EhV-203, EhV-207 and EhV-208 like; and D=EhV-84 and EhV-86 like). The optimal tree with the sum of branch length = 0.08538966 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (10, 000 replicates) is shown next to the branches. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site.
-
The amplified region of the SPT gene targets the most variable section of this gene among laboratory isolates and corresponds to the linker region between the two domains of the SPT protein: LCB1 and LCB2. Based on the full genomic sequences available, we modelled the entire SPT from EhV-99B1 and EhV-86 (Fig. 5). 89% of the final model of the SPT protein was modelled with an accuracy of >90%, based on the following seven protein structures on the Protein Databank in a decreasing order of alignment coverage and % of confidence: 3LWS, 1BS0, 3HQT, 3A2B, 2BWN, 3TQX, 2W8W. We then incorporated the 'new' environmental fragment sequences into the EhV-99B1 full version, essentially replacing the EhV-99B1 linker region with environmental linker regions, and predicted their theoretical structures.
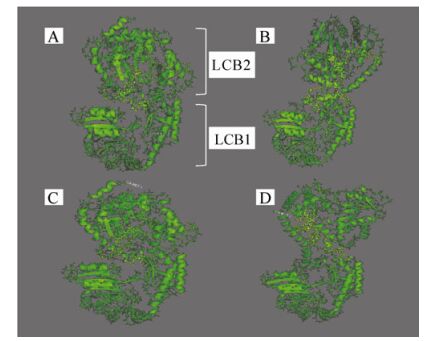
Figure 5. 3D models of four different hypothetical structures of the SPT protein. A) laboratory strain EhV-99B1 isolated originally from a Norwegian Fjord, B) laboratory strain EhV-86 isolated originally from the English Channel, C) sequence from the DISCO cruise in the North Sea, and D) sequence from the L4 sampling station in the English Channel. In yellow are highlighted the amplified regions between the two domains LCB1 and LCB2.
From the secondary structure prediction (Supplementary material Fig. S5) and the 3D models it was clear that the linker region of the SPT protein has the potential to differ structurally among strains. The two domains LCB1 and LCB2 of the protein are potentially closer to each other in EhV-99B1 than in EHV-86 (Fig. 5) and this was most likely due to two amino acid changes in the linker between the two domains; i.e. in EhV-86 Tyrosine and Glutamic Acid were both replaced by Aspartic Acid. The changes from Tyrosine to Aspartic Acid affected the position and the length of the first alpha helix in the sequenced region, resulting in a shorter helix in EhV-99B1 (Supplementary material Fig. S5). In addition there were also potential differences between the modelled fold of L4/16.09.2002 and L4/15.07.2003, and the DIS/23.06. 1999/100m and DIS/29.06.1999/80m sequences. The first alpha helix in the L4/16.09.2002 and L4/15.07.2003 structures was predicted to be longer than the same helix in EhV-99B1, EhV-86, DIS/23.06.1999/100m and DIS/29.06. 1999/80m structures (Supplementary material Fig. S5). The helix was 11 aa long and had an altered position. This was most likely a result of an aa change immediately after the helix, i.e. Proline, Serine and Tyrosine instead of Proline, Serine and Aspartic Acid. The sequence PSY has a net neutral charge while the sequence PSD is negatively charged. Regardless of this, the predicted folds of the L4/16.09.2002 and L4/15.07.2003 derived sequences were more similar to the fold of the EhV-86 SPT than the fold of EhV-99B1, and the folds of DIS/23.06.1999/100m and DIS/29.06.1999/80m structures were more similar to the fold of the EhV-99B1 than the EhV-86 version (Fig. 5).
-
Having identified a potential structural difference in a protein known to be crucial during coccolithovirus infection, we then assessed if such changes are associated with phenotypes of ecological relevance. In order to determine if coccolithovirus genetic diversity can be translated into functional diversity, we determined the simple infection kinetics of two strains with distinct SPT genotypes. Following the inconsistent lysis of Emiliania huxleyi CCMP 2090 by EhV-99B1, we compared the infection kinetics of EhV-86 to those of the more reproducible EhV-V1. EhV-V1 is thought to be almost identical to EhV-99B1 (which itself displays 96.5% pairwise nucleotide identity to EhV-86) having been isolated from the same location and date (Allen M J, et al., 2007; Pagarete A, et al., 2012). Genomic analysis has revealed identical SPT and MCP sequences for EhV-99B1 and EhV-V1. The rate of E. huxleyi cell lysis was higher in the EhV-86 infected cultures than in those infected by EhV-V1. Following infection, the initial number of free VLPs in the first day post-infection differed in that there were fewer free EhV-V1 VLPs than EhV-86 VLPs in the infected cultures (Fig. 6). Then, 48 h later (3 days postinfection) we observed a rapid increase in free EhV-86 VLPs, not seen in the EhV-V1 infected cultures, an indication perhaps that the EhV-86 strain was replicating and releasing virions rapidly, whilst EhV-V1 was much slower with a more steady replication cycle and virion production. It was not until the fifth day after infection that free EhV-V1 VLPs were observed in higher numbers. However, 9 -12 days post-infection, the amount of free VLPs of both viruses were similar, at around 5 × 108 VLP mL-1.
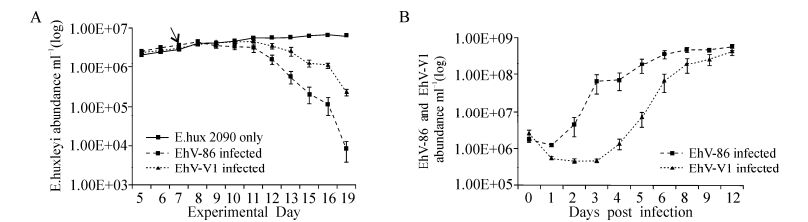
Figure 6. A: E. huxleyi CCMP 2090 average growth mL-1 (of five replicates) following infection by EhV-86 and EhV-V1. The cultures were at a host density of ~2×106 mL-1 at the time of infection and the virus was added in a ratio of 1:1 with the host on day seven (indicated by the arrow). The cell density; B: The free VLPs were enumerated using AFC (Red and Green fluorescence vs side scatter). No virus was added to the control cultures hence the plot was below the detection level and is not seen in B.
Environmental Sampling
Coccolithovirus Major Capsid Protein Diversity
Structural Implications of MCP Diversity
Coccolithovirus SPT Diversity
Structural Implications of SPT Diversity
Infection Dynamics
-
Previously, studies that have looked at natural populations of coccolithoviruses and/or their infection kinetics have relied upon the detection of VLPs via direct enumeration by AFC in combination with molecular fingerprinting where applicable (Martinez J M, et al., 2012; Martinez J M, et al., 2007). However, these studies depend on high numbers of host and their associated virus community. Here, with the exception of the samples collected during the DISCO cruise, we have concentrated on environmental samples with no obvious coccolithophore or coccolithovirus populations present at the time of sampling. Until now, there has been little if any information on the "invisible" abundance of coccolithoviruses under non-bloom conditions, a state in which the host is found during the majority of its life cycle in the world's oceans. There was strong molecular evidence to suggest that coccolithoviruses were present along the entire Atlantic transect, adding further support to the 'everything is everywhere' theory (de Wit R, et al., 2006).
Proven and established molecular markers such as the MCP gene are useful when the aim is to study natural samples where the preliminary information about the sites of sampling and potential habitat range of the host is limited. However, MCP is limited when attempting to make functional inferences based directly on the phylogenetic diversity measurements. Here, we developed a new genetic marker and attempted to make such functional inferences. While the SPT marker lacked the sensitivity and/or spatial coverage of the established coccolithovirus MCP marker, we did manage to infer structural (and thus potentially functional) differences based on the information obtained from using it. It is interesting to note the lack of amplification of the SPT fragment from any of the AMT samples. The failure to detect the SPT gene from the AMT samples is likely a consequence of low detection limits, in that the primers just may not be sensitive enough for low abundance templates. Indeed, the L4 and DISCO samples showed a consistently lower rate of successful amplification of a PCR product for SPT than MCP. Alternatively, the coccolithovirus SPT gene could be absent entirely or present in a more divergent form. To date, all coccolithoviruses that have been isolated and sequenced have been shown to harbour the SPT gene, regardless of their geographical source of isolation (Allen M J, et al., 2006; Nissimov J I, et al., 2011; Nissimov J I, et al., 2011; Nissimov J I, et al., 2012; Nissimov J I, et al., 2012; Pagarete A, et al., 2012; Wilson W H, et al., 2005). However, the gene is the product of a horizontal gene transfer event from the host, E. huxleyi, and is unique in being the only single-chain SPT biochemically characterised and comprises a direct fusion of the LCB1 and LCB2 domains (Han G, et al., 2006; Monier A, et al., 2009). Since the target of the primers is the linker region between these domains, SPT may still be present in the AMT coccolithovirus community, but in a two component system yet to be linked up. Indeed, during the characterisation of the EhV-86 SPT, the co-expression of the individual LCB1 and LCB2 domains could rescue SPT deficient yeast mutants (Han G, et al., 2006). We have recently isolated a new coccolithovirus, tentatively called EhV-18, in which this appears to be the case (Nissimov and Allen, unpublished). Full genomic sequencing is currently underway to confirm this discovery.
However, as we have shown here, 3D modelling of the coccolithovirus SPT protein suggests that the conformation of the two domains has the potential to be altered when the linker section is different, even if this difference is a change in only two or three amino acids. LCB2 and LCB1 are potentially further away from each other in the EhV-86 encoded SPT than in the EhV-99B1 version of the predicted protein, and further apart still in some of the environmental isolates. SPT is the first and rate limiting step in the sphingolipid biosynthesis pathway (Han G, et al., 2006; Michaelson L V, et al., 2010). The distance between, for example, the pyridoxal phosphate binding lysine located on the LCB2 domain and the cysteine residue in the glycine motif on the LCB1 domain, may be crucial in determining the rate and speed of co-factor binding. If this is indeed the case, variations in the amino acid sequence between the two domains could dramatically influence the rate of this reaction, and as a consequence the rate of the infection cycle. Indeed, previous expression of the separated domains showed an active SPT enzyme displaying similar properties, albeit it with a reduced activity, in comparison to the wild-type fusion protein, suggesting structural differences in the linker region could have an impact on enzyme kinetics (Han G, et al., 2006).
Indeed, our identification of clear infection dynamic differences between distinct coccolithovirus strains suggests that 'similar' virus strains can differ with regards to at least one aspect of the infection or/and replication cycle. Clearly, we can make no firm conclusions as to the exact genetic or metabolic basis for the differences observed. However, the observation that ultimately there was an equal amount of virions produced by both virus isolates is interesting: both viruses have the same productivity potential, although one differed in its rate of production. A likely candidate is therefore, the rate limiting SPT enzyme in a pathway that has been suggested to be of crucial importance to successful infection and host cell fate (Bidle K D, et al., 2011; Bidle K D, et al., 2007; Pagarete A, et al., 2009; Pagarete A, et al., 2011; Vardi A, et al., 2009; Vardi A, et al., 2012). The implications of this could be important with regards to biogeochemical cycling in the oceans where nutrients are being constantly recirculated via the "microbial loop" and "viral shunt" into different trophic levels and carbon export to the deep ocean following viral-induced E. huxleyi bloom termination (Brussaard C P, et al., 2008; Falkowski P G, et al., 2008; Suttle C A, 2005; Suttle C A, 2007).
Even though we have not attempted an extensive analysis to link environmental parameters with our molecular data in this study (future more comprehensive studies will need to be undertaken), we have made a few ecologically relevant observations. Although, the method used here for capturing DNA ensured we could not distinguish between free drifting coccolithoviruses and those attached to or within cells, the observation that the number of distinct MCP bands increased with increasing depth, raises interesting questions of the strategy of this group of viruses to "survive" in times of reduced activity and rate of infection. When distances between host cells are small (i.e. high host density), viral infection is rife and the host population can be rapidly decimated, as seen under E. huxleyi bloom conditions (Schroeder D C, et al., 2003). However, viruses must survive the lean times when host numbers are sparse. Exposed to, and at the whim of, the environment, viruses will quickly perish. A possible mechanism for preventing this could be their association with the deep chlorophyll maximum (DCM) region, where the typical percentage of light penetrating is around 1% and the infection and virus production turnover is likely somewhat slower. The depths could harbour the diverse reservoirs of genetic potential exploited under bloom conditions overhead (when virus abundances are high, but their relative diversity is low), in an 'infection from below' dynamic. Such a hypothesis can be supported by the observation that most samples from the DCM region at the AMT-20 transect had a larger number of distinct MCP sequences than the samples from shallower depths, and thus a higher diversity of coccolithovirus genotypes. In conclusion, the use of phylogenetic markers such as the MCP gene are useful in the mapping of coccolithoviruses on a spatial scale, and the investigation of genotype diversity on a temporal scale, even when the number of virus particles is low and their detection impossible by conventional techniques. However, an investigation of a conserved gene for phylogeny cannot reveal much about the functional characteristics of a given community of viruses or particular strains. Therefore, additional markers such as the SPT gene can be used in conjunction with the MCP marker to not only provide additional diversity information, but also provide crucial insights into the function and metabolic potential of the viruses under study. Moreover, an investigation of functionally important genes and their 3D theoretical modelling can provide an additional insight into the importance of these genes, their possible mode of operation, and raise new hypotheses that can be experimentally tested in the future. The results here provide further evidence that, the often ignored, intra-family biodiversity could have real and measurable impact on community and ecosystem composition of ecologically important groups of marine viruses.
The role of SPT as a key component of the sphingolipid pathway during coccolithovirus infection of the coccolithophores, has gained increasing attention in recent years (Bidle K D, et al., 2011; Bidle K D, et al., 2007; Han G, et al., 2006; Michaelson L V, et al., 2010; Pagarete A, et al., 2009; Vardi A, et al., 2009; Vardi A, et al., 2012). Implicated in both cellular signalling and structural roles, control over sphingolipid production appears crucial during infection. Previously, we have shown a distinct battle to control the expression of the sphingolipid pathway occurs under natural bloom conditions (Pagarete A, et al., 2009). Here, we have identified a potential structural difference in SPT, and determined that strains harbouring distinct SPTs do indeed display different infection phenotypes. However, whether the phenotypes displayed are related directly to SPT function could not be determined in this study, and the evidence at this stage is merely circumstantial. The genetic manipulation of virus genomes in the future will hopefully allow these questions to be addressed. For now, we must be content with the knowledge that coccolithovirus diversity exists, this diversity can be measured within the natural environment and, crucially, this genetic diversity is translated into environmentally relevant phenotypes, contributing to and impacting upon global ecosystem function. This study represents the start of the journey to link coccolithovirus biodiversity measurements to their wider role in ecosystem function.
-
This work was funded by the NERC Oceans 2025 program, Plymouth Marine Laboratory's Research Program and the annual AMT program co-organised by the Plymouth Marine Laboratory and the National Oceanographic Centre in Southampton. We would like to acknowledge the NERC National Capability funded Western Channel Observatory (www.westernchannelobservatory.org.uk) which enables the sampling at L4 to take place. We would also like to acknowledge Dr Willie Wilson and Dr Joaquin Martinez-Martinez for collecting and archiving the DISCO samples which were re-analysed in this research article. JIN is a NERC funded PhD student.
-
JIN was responsible for the collection of the samples from the field, extracting the DNA samples, conducting the molecular work in the laboratory, the computational analysis, and the initial drafting of the manuscript. MJA helped in the extraction of the field DNA samples and helped with the molecular work, and SAK took part in conducting the infection dynamics experiment and analysis of the Flow Cytometry samples. The project was co-supervised by MJA, SAK, JAN and CBN and all authors took an active part in the initial design of the experiments, data interpretation, the writing and approval of the final manuscript.
-
The supplementary materials are available on the website of Virologica Sinica: http://www.virosin.org.


 DownLoad:
DownLoad: