











HTML
-
Varicella zoster virus (VZV), upon encountering a naïve host, causes a primary infection commonly known as chickenpox (varicella) (Abendroth A, et al., 1999; Arvin A M, 2001). The disease is generally considered as mild and self-resolving even in the absence of treatment (Arvin A M, 1995), although occasionally it has severe and lethal consequences (Choo P W, et al., 1995; Gilden D H, et al., 2000). The virus reaches sensory nerve ganglia where it remains latent for life, unless temporary or permanent immune suppressive conditions within the host facilitate its reactivation as shingles, or herpes zoster (HZ), affecting thoracic, cranial or lumbosacral dermatomes. The reactivation is sometimes associated with postherpetic neuralgia (PHN), a severe pain along the affected sensory nerves that can linger for years even after the herpetic rash resolves (Arvin A M, 2001; Drolet M, et al., 2010; Liesegang T J, 2004; Lydick E, et al., 1995; Opstelten W, et al., 2002). The drug treatments available to date against VZV elicited diseases are only useful in alleviating some of the symptoms and in shortening the disease duration, but cannot clear the virus or prevent establishment of latency (Hatchette T, et al., 2008; Klassen T P, et al., 2005). PHN is difficult to manage especially in the elderly, who frequently suffer from other age-related conditions and the use of the standard PHN treatment including tricyclic antidepressants, anticonvulsants and opioids can be hazardous (Drolet M, et al., 2010; Lydick E, et al., 1995).
-
In 1995, the live attenuated varicella zoster virus (VZV) vaccine was approved for use in the United States (Uebe B, et al., 2002). Most children receive the first of a two-part vaccination series at their twelve-month pediatric visit. The VZV vaccine has been shown to be approximately 90% effective (Uebe B, et al., 2002). However, the initial live attenuated VZV vaccination may establish a latent infection, which can cause herpes zoster (HZ) upon reactivation (Chesnut G, et al., 2012; Uebe B, et al., 2002; Vazquez M, 2004). Herpes zoster is common, with an estimated one million new cases annually in the United States. The incidence after age 60 is 8-to-10-fold greater than in people under 60 (Harnisch J P, 1984). As the US population ages, the incidence of zoster-associated morbidity and mortality will increase. Furthermore, 40-45% of zoster patients over age 60 develop postherpetic neuralgia (PHN), pain that persists more than 4-6 weeks after acute zoster. PHN is the most common complication of zoster. Some zoster patients also develop stroke from large vessel granulomatous arteritis, multifocal infarcts from diffuse small vessel arteritis, myelitis, zoster paresis, and even pain without rash (zoster sine herpete). Zostavax, the currently available HZ vaccine, is simply a higher titer live attenuated v-Oka inoculum, which is only 51% effective in lowering the risk of HZ in the elderly, and is contraindicated during pregnancy and for those with active infections or with extremely weakened immune systems (Kimberlin D W, et al., 2007). A worrisome rise in shingles occurrence in young healthy adults in the post-vaccination era warrants doubled efforts to prevent this potentially debilitating disease (Leung J, et al., 2011; Whitley R J, 2005; Yih W K, et al., 2005).Additionally, the v-Oka vaccine is far from fully effective in preventing the initial varicella disease, as breakthrough varicella is not uncommon and occurs in 15% to 20% of vaccinated patients (Chesnut G, et al., 2012). In addition to children developing breakthrough varicella, the incidence of HZ in vaccinated children is approximately 14 cases per 100,000 person-years (Uebe B, et al., 2002). The cause of HZ in vaccinated children is controversial, but may relate to reduced immunogenicity of the varicella strain used in the vaccine (Uebe B, et al., 2002). Neurological complications of zoster are among the foremost problems, including post herpetic neuralgia, myelitis and central nervous system vasculopathy, possibly leading to stroke. One out of three people in the USA will develop shingles and there are an estimated one million new cases each year. Therefore, it is essential to understand how VZV establishes and reactivates from latency in ganglia, and how the host immune system controls primary, latent and reactivated VZV infection. A better understanding of VZV pathogenesis and neuroinvasion will lay the groundwork for a neuro-attenuated vaccine that will circumvent all disease caused by VZV in the yet-to-be vaccinated, or exposed, populations.
-
VZV is fundamentally different from the simplex viruses and unique in the herpes paradigm. For instance, VZV has a strict cellular association in cultured cells and is almost entirely human host restricted. These qualities posed many challenges in the efforts to characterize its genes and set it apart from the other alphaherpesviruses, whose host promiscuity and capacity to generate sufficient amounts of cell-free particles allowed for a much earlier understanding of their replication and pathogenesis. Previous methods of creating recombinant VZV deletion mutants utilized the four-cosmid system, in which VZV DNA was split into four overlapping segments called cosmids. In this system, one cosmid would be made to contain the desired mutation, and all four segments are then co-transfected into human cells to produce the recombinant VZV variant upon multiple homologous recombination events (Cohen J I, et al., 1993; Niizuma T, et al., 2003). This method, however, was inefficient as it required co-transfection of four large cosmids into human cells and multiple successful homologous recombination events within a single cell to create the fulllength viral genome (Nagaike K, et al., 2004). Fortunately, building upon the overlapping cosmid system, the bacterial artificial chromosome (BAC) technology yielded a new and more efficient method for generating recombinant viral mutants. BACs are very useful due to their low copy number and the ease of producing mutants with the advantage of harboring them in modified E. coli with λ prophage homologous recombination machinery. Insertion of a BAC vector into one of the genomic cosmids via homologous recombination is the key step that allows for the rapid creation of recombinant viruses that can be genetically manipulated in E. coli. In order to circumvent some of the problems in the study of VZV with cosmids, four cosmids (pvFsp73, pvSpe14, pvPme19, and pvSpe23) with overlapping genomic sequences of VZV were created (Figure 1A). The BAC vector (pUSF-6) containing the origin of bacteria replication and selectable markers is inserted in between VZV ORF60 and 61 in the pvSpe23 cosmid (Figure 1B) (Warden C, et al., 2011). The overlaps between cosmids allow for homologous recombination between the segments and formation of a single, circular DNA sequence that encompasses a full-length infectious VZV genome. Viral replication and plaque formation can be visualized due to the GFP marker in the BAC vector.

Figure 1. Construction of VZV BAC and VZV Luciferase. A: Schematic diagram of the VZV p-Oka genome. Image adapted from Zhang, et al. (2007), including four cosmids containing overlapping VZV genomic segments. A BAC vector (pUSF-6, shown in green) was inserted between ORF60 and ORF61 in VZV cosmid pvSpe23 by homologous recombination. Image adapted from Zhang, et al. (2007). B: All four VZV cosmids were transfected into MeWo cells (1), and upon multiple successful homologous recombination events (2), the resulting recombinant virus was able to replicate and produced a green fluorescent plaque (3). The VZV BAC DNA was extracted from the cells (4)and transformed into electrocompetent DY380 E. coli in the presence of chloramphenicol (5). The infectivity of the recombinant BAC is tested by extraction from the E. coli (6) and transfecting it into human MeWo cells (7). The VZV BAC produced GFP-expressing virus that formed plaques (8). C: To generate a VZVLUC strain, a luciferase expression cassette was inserted into the intergenic region between ORF65 and ORF66 of the VZVLUC genome. This clone was then transfected into MeWo cells to produce the VZVLUC strain. D: MeWo cells were grown in six-well culture dishes and infected with VZV BAC or VZVLUCBAC. Bioluminescence from VZVLUC infected cells was then visualized and measured using IVIS (Xenogen) imaging. Strong bioluminescence was detected only from VZVLUC-infected cells. In addition, green fluorescent plaques were observed for all wells using fluorescence microscopy, indicating that both viruses were infectious.
However, it is near impossible to quantify the amount of GFP expressed to accurately analyze the growth kinetics of viruses, especially when an animal model is used. For this reason, bioluminescent imaging (BLI) has become a powerful tool for studying herpesviruses in vivo and in vitro (Contag C H, et al., 1997; Rehemtulla A, et al., 2000). BLI is a novel method that enables the monitoring and measurement of viral replication in live cells, tissues and animals. The method involves the use of reporter genes, such as luciferase, that are inserted into the viral genome and expressed only as viral proteins are expressed during replication (Hastings J W, 1983; Kurfurst M, et al., 1983). When exposed to the D-luciferin substrate, luciferase will catalyze the substrate’s bioluminescent oxidation, thus producing light whose intensity is dependent on the amount of luciferase present (Doyle T C, et al., 2004; Hastings J W, 1983). BLI has several advantages over other imaging systems such as fluorescent-based imaging. Firstly, BLI is especially effective for in vivo applications because D-luciferin can rapidly permeate the tissues of a living animal, allowing the substrate to reach any site of infection in the body, and therefore poses no limits for in vivo applications.
Additionally, in comparison to GFP, the shorter wavelength of the photons emitted from the luciferaseluciferin reaction allows the light to penetrate tissues and return to the detection instrument for accurate quantification. D-luciferin also has a low toxicity that allows the substrate to be used repeatedly in the same animal (Contag C H, et al., 1997; Rehemtulla A, et al., 2000). Above all else, the main advantage of BLI over other methods is that it can monitor viral growth and provide real-time detection of spatial and temporal growth of viruses in vivo. This makes quantification of viral growth in vivo possible, an attribute necessary for analyzing recombinant viral growth kinetics and tissue tropism (Collaco A M, et al., 2005; Contag C H, et al., 2002; Fan S, et al., 2005; White M R, et al., 1995). Coupling VZV BAC technology with BLI by adding a luciferase gene into viral DNA offers even more advantages for understanding VZV pathogenesis. A luciferase-expressing virus can be used to detect areas of viral infection and, more importantly, study the growth kinetics of recombinant mutated viruses and interpret the resulting data to identify tissue-tropic genes. Furthermore, using a bioluminescent-capable VZV facilitates studies of anti-VZV compounds, VZV pathogenesis and mutational analyses. The first step to generate a replication competent VZV with a luciferase marker (VZVLUC) is to insert a firefly luciferase expression cassette into the intergenic space between ORF65 and ORF66 (Figure 1C) (Zhang Z, et al., 2007). This DNA is then transfected into ARPE-19 cells (a human retinal pigment epithelial cell line) to create the VZVLUC virus. Bioluminescence is only detected in VZVLUC-infected cells (Figure 1D).
-
Functional analysis of each ORF can be performed by mutational analysis of deletion mutants. PCR methods are employed to replace each ORF with a selectable marker via homologous recombination in bacteria and the resulting BAC clone can be transfected into human cells for virus propagation and mutational analysis. Despite having the smallest genome of the eight herpesviruses, not even half of VZV’s ORFs have been characterized previously (Zhang Z, et al., 2007). Creating deletion mutants using the aforementioned methods has allowed for functional studies of VZV’s 70 unique ORFs. Understanding the roles and essentiality of each ORF is the first step to identify VZV tissue tropic factors, which will contribute to the development of a skin- and neuroattenuated vaccine.
The method for generating VZV mutation clones and the corresponding rescue viruses is the galK-based mutagenesis selection/counterselection system. The galK-based mutagenesis also relies on the homologous recombination system mentioned above, but this system possesses an advantage over an antibiotic selection system in that no foreign DNA will remain in the rescue virus. The E. coli SW102 strain is used because the galK gene of the galactose operon in these bacteria is defective. As a result, the strain cannot survive in a medium where galactose is the only source of carbon. This system supplies the galK gene in trans (within the BAC) so that the bacteria will only grow when a homologous recombination event replaces ORFX with galK (Warming S, et al., 2005; Zhang Y, et al., 2000; Zhang Z, et al., 2008).
Using the aforementioned methods, a library of all 70 ORF deletion mutants was created and screened in melanoma cells and skin organ cultures (Zhang Z, et al., 2010), in an effort to reveal all the genes involved in skin tropism. In addition to previously known tissue tropic genes, we found ORF7 to be the single novel gene whose absence prevented VZV replication and spread in skin organ culture ex vivo and in an in vivo SCID-hu mouse model. A schematic of the process for making the ORF7S (where 3 stop codons have been inserted into the ORF7 gene), is outlined in Figure 2.
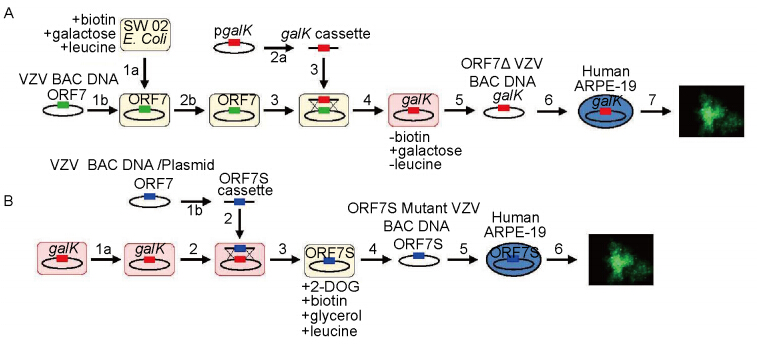
Figure 2. VZV ORF7 mutational analysis and ORF7S mutant generation by galK-based mutagenesis system. A: Generation of ORF7 deletion mutant virus. SW102 E. coli are made to be electrocompetent (E.C.) (1a) for VZV BAC DNA transformation (1b). Next, a galK gene is PCR amplified by primers containing 40-bp sequences homologous to the flanking regions of ORF7 in VZV to create the galK cassette (2a). In order to generate a deletion mutant clone (ORF7Δ), SW102 E. coli carrying WT VZVBAC undergo E.C. cell preparation with recombinase system activated (2b). The galK cassette is then electroporated into the E.C. SW102 harboring WT VZVBAC (3). Upon homologous recombination, ORF7 will be replaced by the galK gene, generating the VZV ORF7Δ mutant BAC clone (4). The deletion clone is then verified by PCR and sensitivity to antibiotics, and extracted via BAC Maxipreparation (5). The mutant BAC is then chemically transfected into human ARPE-19 cells (6)and grown for mutant analysis (7). B.Generation of ORF7S mutant VZV virus: SW102 E. coli harboring VZVLUC ORF7Δ mutant BAC is made electrocompetent and recombination system activated (1a). A previously constructed VZV ORF7 with stop codons inserted in the 5’ end of the gene (ORF7S) is PCR amplified from BAC DNA template with primers conferring homology to flanking regions in ORF7Δ mutant BAC (1b), so that upon transformation (2) and homologous recombination (3), the ORF7S cassette will replace the galK gene in the ORF7Δ mutant BAC. After PCR verification, growth property confirmation, and extraction of the rescue/mutant BAC via Maxipreparation (4), the ORF7S Mutant BAC is chemically transfected into human ARPE-19 cells (5). The virus that results is then grown and analyzed for comparison to WT VZV (6). *Images adapted from Dulal K, et al. (2012).
The essentiality of each ORF is determined by transfecting ARPE-19 cells with mutant DNA and observing viral replication and plaque growth. Through the systematic deletion of each ORF and the transfection of mutants into ARPE-19 cells, the essentiality of each ORF can be determined. If the ORF is nonessential, a viral plaque will be observable within 3–5 days postinfection. A viral plaque that is much smaller than that of a WT infection suggests that the respective ORF strongly influences growth. If no plaque formation is observed, the ORF is likely to be essential for viral replication. A color-coded map of the VZV genome is presented in Figure 3 to represent the results of the global scale mutational analysis of the individual ORF essentiality for viral replication.
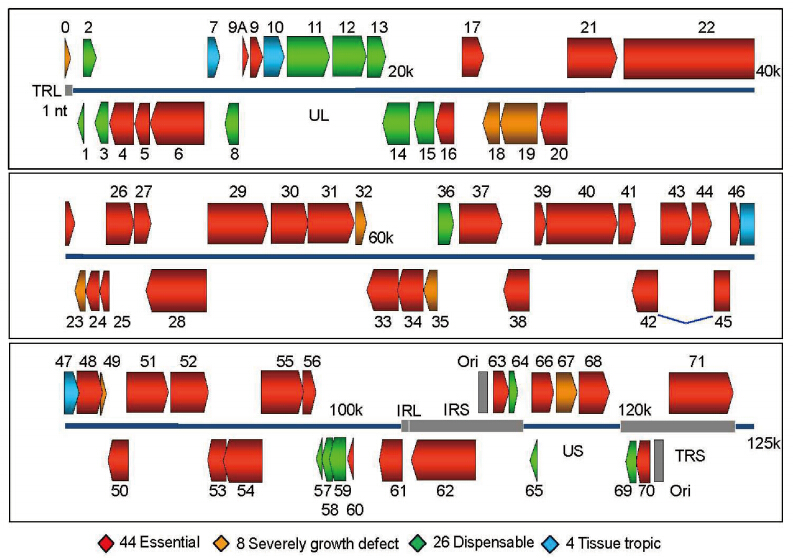
Figure 3. Map of VZV open reading frames organized by essentiality and tissue tropism. Genomic organization and ORFs arrangement are based on the viral sequence of the VZV pOka strain (parental strain of WT VZV). Each VZV ORF is color-coded according to the growth properties of its corresponding virus gene-deletion mutant in cultured MeWo cells and human fetal skin organ cultures. The blue lines for ORF42 represent a splicing junction. For all growth curves, wild-type infections served as positive controls and mock infections served as negative controls. *Image adapted from Zhang Z, et al. (2010)
Subsequent to the identification of a novel skintropic factor, we sought to identify any potential neurotropic factor of VZV by screening the deletion mutants deemed dispensable for viral replication in culture. These genes were targeted due to their capacity to grow without significant defect in culture, with a strong likelihood that one of the genes would be responsible for or involved in tissue tropism, as was seen for the skintropic screening. It was hypothesized that any identified genes that prevent growth or spread in neural models, while remaining replication competent in epithelial or fibroblast cells, would therefore be potential candidates for a live-attenuated vaccine against latency establishment and herpes zoster.18 ORF deletion mutants were transfected, re-spectively, into differentiated neuroblastoma (SHSY5Y) cells and hESC-derived neurons. 17 ORF deletion mutants grew normally in these neuronal models, while only ORF7 deletion mutant proved to be a neurotropic factor, in that the virus failed to replicate and spread in both SH-SY5Y and hESC-derived neurons. In addition, ORF7 deletion mutant VZV was screened ex vivo and in vivo in fetal dorsal root ganglion models (SCID-DRG). The SCID-hu DRG model has proven an excellent tool for the study of VZV pathogenesis and latency in human neurons. Moreover, factors implicated in neuropathogenesis are being brought to light by employing this model. Zerboni et al. have also demonstrated that the live attenuated VZV vaccine strain has similar replication properties to the clinical VZV isolates, which correlates with our results in differentiated neuroblastoma cells (Zerboni L, et al., 2011; Zerboni L, et al., 2010). Our work led to the characterization of ORF7 as the first, and currently only, known neurotropic factor of VZV. Of special interest is the fact that an ORF7 deletion mutant virus can prevent viral spread in skin and in neurons, the two tissues of VZV disease manifestation. Figure 4 presents VZV growth curves for ex vivo SOC (A) and in vivo SCIDhu DRG (B) to demonstrate the different models for studying VZV and the tissue tropism of ORF7.
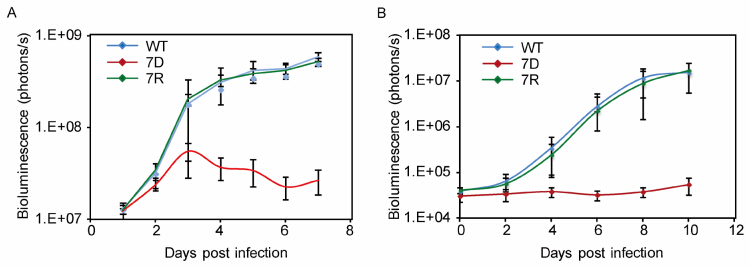
Figure 4. VZV ORF7S (7D) growth kinetics in skin organ culture and dorsal root ganglion. A.Ex vivo growth curve analysis of ORF7D VZVLUC in human fetal skin organ cultures (SOC). Skin tissues were inoculated with 5×103 PFU of either WT VZVLUC, ORF7D VZVLUC, or ORF7 rescue (ORF7R) VZVLUC viruses, in parallel.VZV replication was monitored daily by IVIS for one week as bioluminescence emitting from each skin culture was measured. Each line represents an average of the data from 3 different skin tissue samples, all infected with the same virus. B. In vivo (SCID-hu mice with DRG xenotransplants) growth curve analysis of ORF7D VZVLUC in parallel with WT VZVLUC and ORF7 Rescue (7R) VZVLUC. The DRG was inoculated with 5×103 PFU WT VZVLUC, ORF7D VZVLUC and ORF7R VZVLUC, as indicated. VZV replication was monitored daily by IVIS for one week as bioluminescence emitting from each DRG was measured. Each line represents an average of the data from 3 different DRG samples, all infected with the same virus. *Images adapted from Dulal K, et al. 2009 and Zhang Z, et al. (2010).
-
ORF7 protein is a virion component of unknown function that localizes to the Golgi compartment within infected cells, suggesting a possible role in efficient viral packaging and egress. ORF7 appears to have a determining function in cell-to-cell fusion and it is likely to work in conjunction with other viral or cellular proteins to achieve this effect. Another possibility might be that ORF7 is important in the process of viral entry into cells, specifically in fusing the viral envelope with the cell membrane. However, it has been speculated that VZV does not favor this mechanism as a mode of entry, particularly in epithelial cells, which explains its almost exclusive cell-association in tissue culture (Gershon M D, et al., 2010). VZV, unlike all other alphaherpesviruses, is confined to the cell body instead of being released into the culture medium by cell lysis, which is an indication that VZV has evolved alternative modes of spread, favoring cell-to-cell rather than virusto-cell spread. It was suggested by Gershon et al. that the cation-independent mannose-6-phosphate receptor (MPRci) present on most VZV permissive cells may confine VZV to a cell associated status by re-routing nascent virions to the late endosome. This pathway is averted in suprabasal epidermis keratinocytes, which lose their MPRci receptors as they mature, thus allowing cell free particle release (Gershon M D, et al., 2010). It is not inconceivable that the cell-associated spread within the host could be a means to evade the immune system during natural infection in order to maintain latency (Cole N L, et al., 2003), in contrast with the cell-free mode of transmission from host to host.
Syncytia formation may be critical for neuroinvasion, thus a polykaryon-defective VZV mutant, such as ORF7D, would be unable to efficiently establish infection of neural tissue. It has been suggested that, within human DRG, VZV infects neurons via fusion of infected satellite cells with the neuronal soma, forming neuronsatellite cell complexes (Reichelt M, et al., 2008; Zerboni L, et al., 2010). Since viral spread within nervous tissue is dramatically impaired, absence of ORF7 protein may trigger loss of infectivity specifically due to the incapacity of the virus to fuse potentially infected satellite cells with sensory neurons; however, further evidence is needed to validate this mechanism. An alternative possibility is that although in epithelial cells ORF7 works to sort virions to the Golgi after the first envelopment event, the situation is different in neurons. Here, secondary envelopment may occur at a different site, as is the case with HSV-1, which acquires its secondary envelope from varicosities and growth cones at the tip of the axons or at axonal mid-distal regions (Saksena M M, et al., 2006). In this case, ORF7 may be essential for sorting to this secondary envelopment site, which remains to be determined.
-
These findings could open multiple avenues for future investigations into the mechanism of VZV neuroinvasion, which will have an important impact upon VZV prevention and therapy. ORF7D VZV, unlike the commonly used vaccine strain v-Oka, is a pure and genetically defined strain lacking ability to infect both human skin and nervous tissue in vivo, thus meeting crucial safety and genetic stability criteria for a next generation chickenpox and shingles neuro-attenuated vaccine. ORF7D could also be used as a safer viral vector for gene therapy and for immunization against other viral or even bacterial antigens. Complete prevention of both varicella and herpes zoster is the only path that will lead to eradication of VZV, and a neuroattenuated, or an otherwise highly immunogenic, VZV vaccine is required.
-
All the authors declare that they have no competing interests. This article does not contain any studies with human or animals subjects performed by any of the authors.


 DownLoad:
DownLoad: