











HTML
-
The predominant route of HIV-1 transmission is via sexual intercourse. During sexual transmission, HIV-1 viral particles or virus-infected cells have to overcome a series of obstacles to establish a successful infection including: mucus; the integrity of the genital or rectal epithelial barriers; host innate defense mechanisms; and the sparsity, activation state and susceptibility of mucosal CD4+target cells (Borrow P, et al., 2010; Derdeyn C A, et al., 2004; Keele B F, et al., 2008; Miller C J, et al., 2005; Temchura V, et al., 2014; Tsai L, et al., 2014; Zhang Z Q, et al., 2004). These obstacles collectively result in a relatively low transmission rate per exposure (Boily M C, et al., 2009; Wawer M J, et al., 2005). However, potential selective advantages acquired by HIV-1, such as enhanced binding to specific cell subtypes, might significantly increase the chances of a successful transmission event.
Recent studies indicate that HIV-1 envelope glycoprotein (Env) gp120 is capable of interacting with integrin α4β7 to potentially increase viral infection (Arthos J, et al., 2008; Cicala C, et al., 2009; Darc M, et al., 2011; Li H, et al., 2011; Nawaz F, et al., 2011; Richardson S I, et al., 2013; Tjomsland V, et al., 2013). α4β7 is expressed on lymphoid cells and mediates cell homing to gut-as-sociated lymphoid tissue (GALT), the dominant site of CD4+ T cell depletion shortly after HIV-1 acquisition and local propagation (Berlin C, et al., 1993; Mehandru S, et al., 2004; Sattentau Q, 2008; Veazey R S, et al., 1998). Lymphocytes expressing activated α4β7 may be preferentially targeted by HIV-1 in GALT, facilitating the swift and profound gut pathogenesis of HIV/SIV infection (Mavigner M, et al., 2011; Wang X, et al., 2009). The close association of α4β7 with primary mucosal portals of HIV-1 infection, specifically genital and rectal mucosa, suggests that α4β7 interaction of viral gp120 may play an role in HIV-1 acquisition and establishment of a successful infection (Cicala C, et al., 2009; Haase A T, 2010; Martinelli E, et al., 2013; McKinnon L R, et al., 2011). However, the bona fide contribution of α4β7 in HIV-1 infection has not been fully clarified. It is shown that a number of gp120s derived from different HIV-1 strains are capable of interacting with α4β7 (Arthos J, et al., 2008; Li H, et al., 2011; Nawaz F, et al., 2011). The majority of previous studies investigating the binding capability of HIV-1 to α4β7 mainly used the recombinant monomeric gp120s. Given that the structure of HIV-1 envelope protein is complex and the monomeric gp120 can be distinct significantly from the native trimeric gp120s anchored in the membrane of HIV-1 virion (Harris A, et al., 2011; Wyatt R, et al., 1998), whether whole HIV-1 virions also possess the α4β7 reactivity is a topic of importance and remains to be further elucidated.
In the current study, after confirming the binding of HIV-1 BaL to various α4β7-expressing cells, we investigated the impact of antagonizing α4β7 on HIV-1 infection of primary CD4+ T lymphocytes.
-
Plasmid pNL4-3 BaL+ containing the backbone of NL4-3 and the Env of BaL (referred to as BaL), TZMbl cell line and antibody Act-1 were from NIH AIDS Research & Reference Reagent Program, Division of AIDS, NIH (Germantown, MD, USA). QT6, a fibrosarcoma cell line derived from Japanese quail, was from American Tissue Culture Collection (ATCC, Cambridge, MA, USA). Integrin expressing plasmids including human pα4, pαE, pβ7 and pβ1, mouse pα4 and pβ7, rat pα4 and pβ7 were amplified from cDNA subtracted from PBMCs of corresponding species using primers listed in Table 1 (Table 1) and inserted into the expression vector pcDNA3.1 (+), respectively. Antibody 2B4 and mouse anti-human IgG were from R & D Systems (Minneapolis, MN, USA). HP2/1 was from AbD Serotec (Oxford, UK). RPA-T4, anti-CD3-PE, anti-CD4-FITC, anti-CD8-FITC, anti-CCR5-PE, anti-human β7-PE, anti-mouse β1-PE, anti-rat β7-PE, anti-mouse IgG-APC and matched isotype control antibodies were from BD Pharmingen (San Jose, CA, USA). Leu3A, anti-human β7-APC and control IgG-APC were from Biolegend (San Diego, CA, USA). Reagents for ELISA assay were from Beckman Coulter (Brea, CA, USA), with a limit of sensitivity of 30 pg/mL.
Name Sequence (5' to 3') Usage α4 forward GAACTAGCTAGCGCATGGCTTGGGAAGCGAGa PCR amplification of human α4 from cDNA α4 reverse GCTCCTGCCTCGAGTCAATTTGAAAGAAGTCCTTAATC β7 forward GAAACACGAATTCTTGGGATCTCGGGCATGGTGG PCR amplification of human β7 from cDNA β7 reverse CCTATTCTAGAGGGTAAGTGTCCCTCCCTCCTTCAGA αE forward GAACTAGCTAGCGCTCCAGCAAGGATGTGGCTCTTC PCR amplification of human αE from cDNA αE reverse TTATGCCTCGAGTCTCCCAGTGGATAGCAGGTCC β1 forward AACGGAATTCAAGATGAATTTACAACCAATTTTCTG PCR amplification of human β1 from cDNA β1 reverse TAGCTCGTCTAGAAGTACTCATTTTCCCTCATACTTC m. α4 forward GACCTAGCTAGCTGTTGAATGTTCTCCACCAAGAGCG PCR amplification of mouse α4 from cDNA m. α4 reverse TATGCCTCGAGGTCTTCAGTCATCATTGCTTTTGCT m. β7 forward AATATGAATTCTGCTCCTCCTCAAGCACCTGCCATG PCR amplification of mouse β7 from cDNA m. β7 reverse CACCTGGTCTAGAACTGTCCTCCAAGACAAGAATCCTAAGTC r. α4forward GACCTAGCTAGCTGTTGAATGTTCCCCACCAAGAGTG PCR amplification of rat α4from cDNA r. α4reverse TTACCTCGAGAGTCTTCAGTCATCATTGCTTTTGCTGT r. β7 forward ACCTAGCTAGCGCCATGGTGGATTCATCAACTGTTC PCR amplification of rat β7 from cDNA r. β7 reverse TTACCTCGAGCTAAGTCAGTCAGCCTCCTGGGTCAG iα4 #1 forward TGCTCCGTGTTATCAAGATTATTTCAAGAGAATAATCTTGATAACACGGAGCTTTTTTG shRNA targeting α4 iα4 #1 reverse TCGACAAAAAAGCTCCGTGTTATCAAGATTATTCTCTTGAAATAATCTTGATAACACGGAGCA iα4 #2 forward TCGGGAGCAGTAATGAATGCAATTCAAGAGATTGCATTCATTACTGCTCCCGTTTTTTG shRNA targeting α4 iα4 #2 reverse TCGACAAAAAACGGGAGCAGTAATGAATGCAATCTCTTGAATTGCATTCATTACTGCTCCCGA iβ1 forward TGCCTTGCATTACTGCTGATATTTCAAGAGAATATCAGCAGTAATGCAAGGCTTTTTTC shRNA targeting β1 iβ1 reverse TCGAGAAAAAAGCCTTGCATTACTGCTGATATTTCAAGAGAATATCAGCAGTAATGCAAGGCA iCCR5 forward TGAGCATGACTGACATCTACTTCAAGAGAGTAGATGTCAGTCATGCTCTTTTTTG shRNA targeting CCR5 iCCR5 reverse TCGACAAAAAAGAGCATGACTGACATCTACTCTCTTGAAGTAGATGTCAGTCATGCTCA scrambled forward TCCTAAGGTTAAGTCGCCCTTTCAAGAGAAGGGCGACTTAACCTTAGGTTTTTTG non-targeting shRNA scrambled reverse TCGACAAAAAACCTAAGGTTAAGTCGCCCTTCTCTTGAAAGGGCGACTTAACCTTAGGA The underlined positions are ezymatic restriction sites, sense or anti-sense sequences of the oligos as implicated. Table 1. Primers and shRNA oligos
-
The complete coding sequences of α4 and β7 were amplified from constructed pα4 and pβ7 and subcloned into the lentiviral vectors pLJM1 (Addgene, Cambridge, MA, USA, with a substitution of CMV-EGFP into CMV-2aRFP) and pLenti6.3/V5-DEST (Invitrogen, Grand Island, NY, USA, with an insertion of a MCS-IRES2-EGFP sequence after the CMV promoter), respectively. The constructed pLJM1-CMV-α4-2a-RFP or pLenti6.3-CMV-β7-IRES2-EGFP/V5-DEST vector was co-transfected with psPAX2 and pMG2.G (Addgene, Cambridge, MA, USA) into 293T cells to produce lentiviruses which were subsequently used to infect CHO cells at optimal multiplicity of infection. Cells with high and stable α4β7 expression were selected by culture in the presence of antibiotics puromycin and blasticidine, and further purified by limited dilution (Wurm F M, 2004).
-
All human blood samples were collected under protocols approved by the Local Research Ethics Committee. PBMCs were isolated from single buffy coats, and stimulated with 20 U/mL interluekin-2 (R & D Systems, Minneapolis, MN, USA)and 1 μg/mL phytohaemagglutinin (Sigma-Aldrich, St. Louis, MO, USA) for 3 days. For α4β7 activation, 10 nmol/L retinoic acid (RA, SigmaAldrich, St. Louis, MO, USA) was added to the medium at the beginning of the 3-day culture, followed by an additional 4-day culture in the presence of IL-2 and RA. CD4+ T cells were prepared from PBMCs by negative selection and cultured as PBMCs (Miltenyi, Bergisch Gladbach, NRW, Germany). CD8+ T cells with high or low/negative α4β7 expression were sorted from activated PBMCs by gating on the CD8+ lymphocytes with corresponding β7 expression.
-
For cell transfectants and CHO-α4β7 cell lines, cells were detached by trypsin and recovered at 37 ℃ for 2 hours. For CD8+ T cells, after sorting by flow cytometry, cells were cultured at 37 ℃ for 2 days prior to binding assay. For each condition, 1 × 106 cell-line cells or 5 × 105 primary cells were incubated with 100 ng p24 of virus at 37 ℃ for 2 hours (for transfectants) or 1 hour (for primary cells) with end-to-end rotation. Inhibitors were present as indicated and pre-incubated with the cells for 1 hour (for transfectants) or 30 min (for primary cells). The incubation medium was supplemented DMEM with the addition of 1 mmol/L MnCl2. After incubation, the cells were washed 3 times using HEPES buffer supplemented with 1 mmol/L MnCl2 and 100 μmol/L CaCl2 to remove unbound virus, lysed using 1% Triton X-100, then subjected to p24 antigen quantification.
-
Short-hairpin RNA interference was carried out as described previously (Qin X F, et al., 2003). In brief, shRNA oligo sequences targeting human integrin α4were downloaded from the RNAi Consortium(http://www.ncbi.nlm.nih.gov/projects/genome/probe/doc/ProjTRC.shtml), analyzed for suitability and potential efficacy according to general guidelines for RNAi design (Birmingham A, et al., 2007; Qin X F, et al., 2003; Tiscornia G, et al., 2003), synthesized, annealed and inserted into the lentiviral vector pLentiLox3.7 (referred to as pLL3.7) (Addgene, Cambridge, MA, USA). Scrambled shRNA and shRNA sequence targeting CCR5 were from Addgene or synthesized as described (Qin X F, et al., 2003) (Table 1), and subsequently inserted into the lentiviral vector pLL3.7. The constructed pLL3.7-shRNA or empty vector were co-transfected with psPAX2 and pMG2.G into 293T cells to produce lentivirus which were then titrated and used to transduce RA-treated CD4+ T cells. 4–6 days post-transduction, the positively transduced cells (GFP+) were analyzed for gene expression by FCM and sorted for downstream virus infection.
-
Pseudotyped reporter viruses were prepared as described (Hu Q, et al., 2005). In brief, 293T cells were co-transfected with plasmids expressing HIV-1 Env or VSV-G and pNL4–3.Luc. R E using Lipofectamine 2000 (Invitrogen, Grand Island, NY, USA) according to the manufacturer's instructions. Infectious HIV-1 was produced by transfecting plasmid BaL into 293T cells using Lipofectamine 2000 according to the manufacturer's instructions. PBMC-originated viruses were produced by infection of PBMCs with 293T-derived viruses. All viral stocks were titrated by p24 ELISA.
For virus infection assays, 2 × 105 PBMCs or CD4+ T cells were infected with 2 ng p24 of virus followed by extensive washes to remove unbound virus. Cells were cultured in supplemented RPMI-1640 for 12 days. Supernatants were collected every 3 days post-infection, lysed and then stored at -80 ℃ until detection for p24 antigen.
-
For expression assays, 5 × 106 cells were used for each condition. In brief, cells were harvested, counted and washed once using PBS with 3% FBS. For integrin transfectants and α4β7-expressing cell lines, cells were washed using HEPES buffer supplemented with 1 mmol/L MnCl2 and 100 μmol/L CaCl2. Cells were stained with indicated antibody for 30 minutes on ice, followed by staining with secondary antibody for another 30 minutes in some cases. The stained cells were washed twice, fixed with 500 μL of 1% paraformaldehyde in PBS and analyzed on a FACSAria Ⅲ (BD, San Jose, CA, USA) cytometer.
For cell sorting, 5 × 107 cells were used for each sample. Cells were stained as described above except that cells were kept in ice-cold washing buffer and immediately sorted after staining.
-
Data are presented as mean ± SD. The difference of mean value was analyzed by the two-tailed student's t test.p < 0.05 was considered statistically significant.
Plasmids, cells, proteins and antibodies
Construction of α4β7 expressing stable cell lines
Preparation of PBMCs and T cell subsets
Virus binding
RNA interference
HIV-1 production, titration and infection
Flow cytometry
Statistical analysis
-
HIV-1 gp120 has been reported to interact with integrin α4β7 expressed on primary lymphocytes and such interaction has been well studied (Arthos J, et al., 2008; Cicala C, et al., 2009; Darc M, et al., 2011; Jelicic K, et al., 2013; Li H, et al., 2011). However, whether gp120-α4β7 interaction mediates the binding of HIV-1 virions to target cells remains to be further clarified (Arthos J, et al., 2008; Etemad B, et al., 2012). We initially performed experiments to examine whether expression of α4β7 alone could mediate HIV-1 binding. We first sought to confirm HIV-1 binding to α4β7 transfectants by cloning and co-expressing human α4 and β7 genes on 293T cells (Figure 1A). HIV-1 BaL bound to 293T cells transfected with α4β7 to a significantly higher level than those transfected with pcDNA3.1. The α4β7-mediated virus binding was dependent upon its level of expression (Figure 1A). These results were confirmed using a second transduced cell line QT6, derived from Japanese quail (Figure 1B). In agreement with previous reports (Arthos J, et al., 2008; Nawaz F, et al., 2011), our data together shown that exogenously expressed α4β7 can mediate HIV-1 binding to α4β7 expressing cells.
α4β7 is a heterodimer comprised of an α and a β subunits. It belongs to the integrin family which includes a large number of members. To date, 18 α and 8 β subunits and 24 different integrin heterodimers have been reported (Abram C L, et al., 2009; Yu Y, et al., 2012). Thus, we assessed whether α4β7 is unique amongst integrins with respect to HIV-1 binding. We cloned two additional subunits αE and β1, which can form αEβ7 and α4β1 heterodimers in vivo, respectively. By transfection of 293T cells, we found that BaL virions also bound to α4β1 transfectants, albeit to a lesser extent, whereas no significant binding was observed on αEβ7 transfectants (Figure 1C). To further identify whether α4β7 mediated virus binding was species specific, we cloned mouse and rat α4β7 and observed that BaL also moderately bound to mouse α4β7, but not to rat α4β7 (Figure 1C). Comparative integrin expression was determined by flow cytometry (FCM) (Figure 1C). The moderate binding to human α4β1 was likely due to the shared subunit and structure similarity between α4β7 and α4β1 (Yu Y, et al., 2012), while binding to the murine ortholog reflects conserved sequence between human and mouse α4β7 (Qi J, et al., 2012; Tidswell M, et al., 1997).
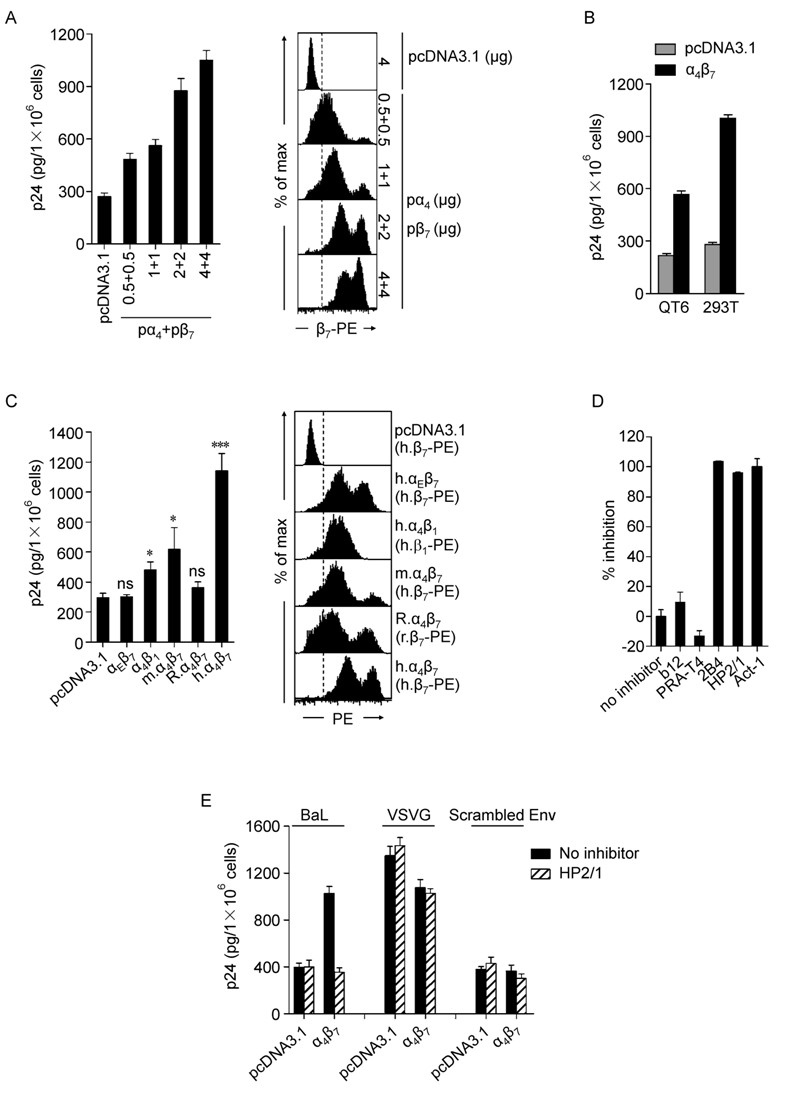
Figure 1. Binding of HIV-1 to cell transfectants mediated by gp120-α4β7 interaction. A-D: 48 hours post-transfection, cells were detached and recovered at 37 ℃ for 2 hours. For each condition, 1 × 106 cells were incubated with 100 ng p24 of HIV-1 BaL in complete DMEM medium with the addition of 1 mmol/L MnCl2 at 37 ℃ for 2 hours with rotation, followed by extensive washes to remove unbound virus. Cells were lysed with 1% Triton X-100 and subjected to p24 measurement. Antibodies (20 μg/ mL) were present as indicated and pre-incubated with the cells for 1 hour. A: Binding of BaL to 293T cells transfected with a serial dose of pα4 + pβ7 or pcDNA3.1 (+) control plasmid (left). The expression of α4β7 was labeled by anti-β7-PE, assayed by flow cytometry and is shown on the right. B: Binding of BaL to 293T or QT6 cells transfected with pα4 + pβ7 or pcDNA3.1 (+). C: Binding of BaL to 293T cells transfected with plasmids expressing human αEβ7, human α4β1, mouse α4β7, rat α4β7, human α4β7, or pcDNA3.1 (+) (left). The expression of the integrins was assayed by flow cytometry and is shown on the right. m, mouse; R, rat; h, human. D: Binding of BaL to 293T α4β7-transfectants in the presence of antibodies to gp120 CD4 binding site (b12), CD4 (RPA-T4), α4 (2B4 or HP2/1) or α4β7 (Act-1). Results are shown as % inhibition, with that in the absence of inhibitors set to 0%. E: Binding of HIV-1 pseudotyped with BaL, VSV-G or frameshiftmutated (scrambled) Env to 293T cells transfected with pα4 + pβ7 or pcDNA3.1, in the presence or absence of anti-α4 antibody HP2/1. All inhititors were presented at 20 μg/mL. Data shown are representative of at least three independent experiments with the bar graphs expressed as mea n ± SD. For virus binding assay, each condition was performed in triplicate. *p < 0.05 and ***p < 0.001, compared to virus binding using cells transfected with pcDNA3.1. ns, not significant.
Subsequently, we measured the effect of inhibitors on virus binding. Results showed that monoclonal antibodies (mAbs, 20 μg/mL) targeting α4 (2B4, HP2/1) or α4β7 (Act-1) almost completely blocked virus binding to α4β7 transfectants, in agreement with a previous report that anti-α4β7 inhibitors abolished gp120-α4β7 interaction (Arthos J, et al., 2008). In contrast, no blocking effect was observed in mock control or in the presence of mAb to CD4 (RPA-T4) or gp120 CD4-binding site (b12) (Figure 1D).
To further explore the specificity of gp120-α4β7 mediated virus binding, we produced pseudotyped HIV-1 using HIV-1 BaL Env, VSV-G or frameshift-mutated Env (scrambled Env), and observed that only BaLpseudotyped HIV-1 bound to α4β7 transfectants and the binding activity was significantly higher than that to pcDNA3.1 transfectants (Figure 1E). The pantropic VSV-G pseudotyped HIV-1 bound to cells regardless of α4β7 expression, whereas scrambled Env pseudotyped HIV-1 lost the capability of binding to α4β7 transfectants. In addition, only binding of BaL to α4β7 transfectants was blocked by HP2/1, an anti-α4 monoclonal antibody (Figure 1E). These results collectively indicate that the interaction between gp120 and α4β7 mediates HIV-1 binding.
-
To further confirm the α4β7-mediated virus binding, we constructed a CHO cell line stably expressing α4β7, using lentiviruses to introduce the α4 and β7 genes into the CHO genome. The expression of α4, β7 and α4β7 on the constructed CHO-α4β7 was analyzed by fluorescence microscopy and FCM (Figure 2A and 2B). Compared to the parental CHO cells, HIV-1 BaL bound to CHO-α4β7 cells at a significantly higher level which could be blocked by HP2/1 (20 μg/mL) (Figure 2C), These results are consistent with those obtained from 293T transfectants (Figure 1).
The above experiments were conducted using cell lines. To assess whether α4β7-mediated HIV-1 binding can be reproduced in primary cells, we carried out virus binding assay using primary CD8+ T cells. This cell subset has little if any CD4 expression, eliminating the interference of CD4 mediated binding. PBMCs were stimulated for α4β7 expression by retinoic acid (RA) (Mora J R, et al., 2003). Activated PBMCs from three different donors were subjected to sorting based on CD8 and β7 expression by FCM (dot plot of Figure 2D). Compared to CD8+ T cells with low or negative β7 expression (β7 low/-), cells with higher β7 expression (β7 hi) mediated markedly higher level of virus binding (Figure 2D). CD4+ T lymphocyte is the primary target of HIV-1 infectionin vivo. After RA treatment, we also observed significant virus binding to primary CD4+ T lymphocytes following blockade of CD4 with mAbs (2.5 μg/mL RPA-T4 plus 2.5 μg/mL Leu3A). This CD4 independent binding was inhibited by the HP2/1 mAb (5 μg/mL) (Figure 2D). These results indicate that α4β7 expressed on primary cells is capable of mediating virus binding.
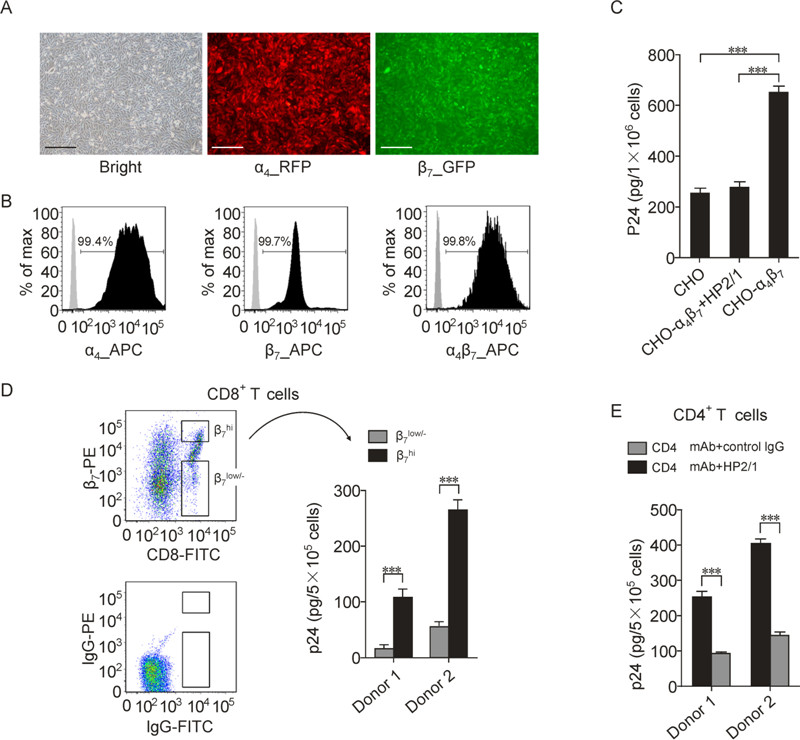
Figure 2. Binding of HIV-1 to a constructed CHO cell line stably expressing α4β7 and RA-treated primary CD8+ and CD4+ T cells. A CHO-α4β7 cell line with stable α4β7 expression were constructed by transducing CHO cells with lentiviral vectors carrying the human α4 and β7 gene, followed by selection using antibiotic resistance and further purification using limited dilution. A: Visualizing of the constructed CHO-α4β7 cells under a fluorescence microscopy. A fluorescent protein RFP or EGFP tag was bicistronically expressed with the α4 or β7 subunit, respectively. The black scale bar represents the length 100 μm. B: Measurement of the α4, β7 and α4β7 expression on CHO-α4β7 cells by FCM. The cells were correspondingly stained with anti-α4 (2B4) and Alexa Fluor 647-conjugated goat-antimouse IgG, or anti-β7-APC, or Act-1 and allophycocyanin-conjugated rabbit anti-mouse IgG, followed by analysis using FCM. C: Binding of HIV-1 BaL to CHO cell lines with or without stable α4β7 expression, in the presence or absence of anti-α4antibody HP2/1 (20 μg/mL). D: Binding of HIV-1 BaL to primary CD8+ T cells with high (β7 hi) or low or negative (β7 low/-) α4β7 expression. The cells were obtained by sorting of retinoic acid-treated PBMCs based on CD8 and β7 expression. The gating strategy is shown by the dot plots. E: Binding of HIV-1 BaL to primary CD4+ T cells. The cells were separated from freshly-isolated PBMCs by negative magnetic selection and cultured for α4β7 activation as PBMCs. The binding experiment was conducted as in described in the legend of figure 1 except that 5 × 105 cells were used for each condition of primary cells. For CD4+ T cells, the cells were incubated with 2.5 μg/mL RPA-T4 plus 2.5 μg/mL Leu3A at 37 o C for 30 minutes to block CD4 receptor and in the presence of HP2/1 (5 μg/mL) to block α4β7 or IgG as a negative control. The bar graphs are expressed as mean ± SD, with each condition performed in triplicate. For C, one out of three independent experiments is shown. For D and E, the experiments are performed using T cells derived from two different donors. ***p < 0.001.
-
Because HIV-1 is capable of binding to primary T cells by interacting with α4β7, we wonder if targeting this integrin could circumvent HIV-1 infection of CD4+ T cells which serve as the main target of HIV-1 infection. Initially, we conducted experiments using anti-α4β7 antibodies. α4β7 expression on CD4+ T cells treated with or without RA was analyzed by FCM (Figure 3A). HIV-1 BaL infection of RA-treated CD4+ T cells was partially inhibited by blocking α4β7 with HP2/1 or Act-1 (5 μg/ mL), while blockade of CD4 with RPA-T4 (5 μg/mL) almost completely inhibited virus infection (Figure 3B). However, anti-α4β7 antibodies could induce cell aggregation (Parrish N F, et al., 2012), with Act-1 being more potent than HP2/1 and the aggregates became remarkably larger when cells were cultured in the presence of higher concentrations of stimulators (i.e., IL-2 plus PHA or OKT3). The phenomenon of anti-integrin induced cell aggregation has been used as a common approach to investigate integrin function as reported by several studies (Andrew D P, et al., 1994; Ruegg C, et al., 1992; Zeller Y, et al., 2001). The formation of cell aggregates increases cell-cell contact, which in turn enhances viral infection (Alvarez R A, et al., 2011; Dale B M, et al., 2013; Liao Z, et al., 2000). This represents an important confounder and suggests that antibody engagement of α4β7 is not an appropriate approach to determine the significance of α4β7 in HIV-1 infection of primary lymphocytes (Parrish N F, et al., 2012).
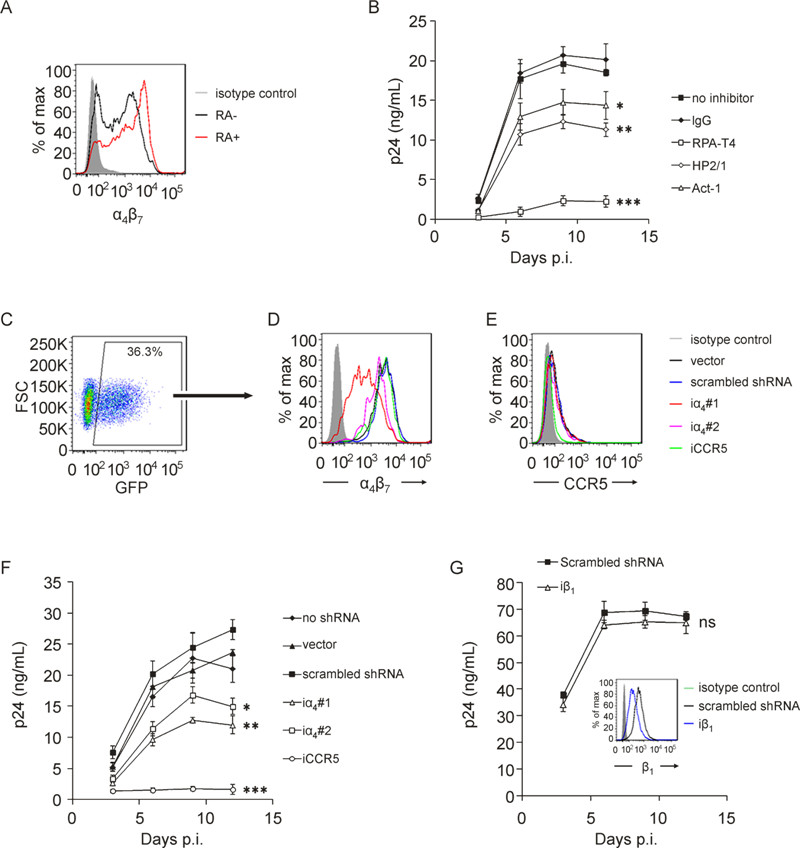
Figure 3. Binding of HIV-1 to a constructed CHO cell line stably expressing α4β7 and RA-treated primary CD8+and CD4+ T cells. A CHO-α4β7 cell line with stable α4β7 expression were constructed by transducing CHO cells with lentiviral vectors carrying the human α4 and β7 gene, followed by selection using antibiotic resistance and further purification using limited dilution. A: Visualizing of the constructed CHO-α4β7 cells under a fluorescence microscopy. A fluorescent protein RFP or EGFP tag was bicistronically expressed with the α4 or β7 subunit, respectively. B: Measurement of the α4, β7 and α4β7 expression on CHO-α4β7 cells by FCM. The cells were correspondingly stained with anti-α4 (2B4) and Alexa Fluor 647-conjugated goat-anti-mouse IgG, or anti-β7-APC, or Act-1 and allophycocyanin-conjugated rabbit anti-mouse IgG, followed by analysis using FCM. C: Binding of HIV-1 BaL to CHO cell lines with or without stable α4β7 expression, in the presence or absence of anti-α4antibody HP2/1 (20 μg/mL). D: Binding of HIV-1 BaL to primary CD8+ T cells with high (β7 hi) or low or negative (β7 low/-) α4β7 expression. The cells were obtained by sorting of retinoic acid-treated PBMCs based on CD8 and β7 expression. The gating strategy is shown by the dot plots. E: Binding of HIV-1 BaL to primary CD4+ T cells. The cells were separated from freshly-isolated PBMCs by negative magnetic selection and cultured for α4β7 activation as PBMCs. The binding experiment was conducted as in described in the legend of figure 1 except that 5 × 105 cells were used for each condition of primary cells. For CD4+ T cells, the cells were incubated with 2.5 μg/mL RPA-T4 plus 2.5 μg/mL Leu3A at 37 o C for 30 minutes to block CD4 receptor and in the presence of HP2/1 (5 μg/mL) to block α4β7 or IgG as a negative control. The bar graphs are expressed as mean ± SD, with each condition performed in triplicate. For C, one out of three independent experiments is shown. For D and E, the experiments are performed using T cells derived from two different donors. ***p < 0.001.
We therefore chose an alternative method, short-hairpin RNA (shRNA) mediated interference, to investigate if disturbance of HIV-1-α4β7 interaction affects HIV-1 infection. A panel of shRNAs were designed and constructed. The sequences of two iα4shRNA candidates, a reported shRNA sequence targeting HIV-1 coreceptor CCR5 (iCCR5) (Qin X F, et al., 2003) and a non-targeting (scrambled) shRNA are shown in Table 1. The expression levels of α4β7 and CCR5 on positively transduced cells (GFP+, 3C) were monitored at various time points post transduction (p.t.). The knock-down effects of two iα4s and the iCCR5 at 4 days p.t.(Figure 3E and 3F) remained constant up to 15 days p.t. (data not shown), consistent with the characteristics of shRNA-mediated interference. iα4 #1 and #2 knocked down α4β7 expression with different efficiency without affecting CCR5 expression (Figure 3D and 3E), indicating that no significant off-target effect was produced. iCCR5 specifically down-regulated CCR5 expression to barely detectable levels (Figure 3E). Lentivirus infection did not cause evident damage to cell viability as measured by trypan blue staining.
The positively transduced GFP+ cells were sorted for HIV-1 infection. Knock-down of α4β7 on CD4+ T cells by both iα4 #1 and #2 significantly reduced BaL infection (Figure 3F). Various efficacies between iα4 #1 and #2 in knock-down α4β7 expression corresponded to an associated reduction in viral infection. Knock-down of CCR5 abrogated HIV-1 infection (Qin X F, et al., 2003), while viral infection was not significantly affected in groups transduced with scrambled shRNA or vector alone, when compared to groups without lentiviral transduction (sorted based on FSC and SSC parameters) (Figure 3F). Additionally, because integrin α4β1, another member of integrin family, was observed to be capable of mediating low level of virus binding (Figure 1C), we determined whether interference of this integrin also affected HIV-1 infection. We found that although introduction of β1-specific shRNA resulted in a notable decrease of β1 expression (the inset of Figure 3G), virus infection of CD4+ T cells was not significantly affected (Figure 3G). Collectively, the above results suggest that both blockade and knock-down of α4β7 expression can decrease HIV-1 BaL infection.
HIV-1 binds to cell transfectants mediated by gp120-α4β7 interaction.
HIV-1 binds to stable α4β7-expressing cell line and primary CD8+ and CD4+ T cells.
Blockade or knockdown of α4β7 expression on CD4+ T cells decreases HIV-1 infection
-
The significance of α4β7 in HIV-1 infection remains controversial. In particular, important concerns have been raised by recent studies regarding whether the ability of some gp120 proteins to engage α4β7 can be recapitulated by HIV-1 virions (Parrish N F, et al., 2012). Our study therefore focused on the impact of gp120-α4β7 interaction on HIV-1 binding and infection of cell line and primary cells. We demonstrated that expression of α4β7 alone is enough to mediate HIV-1 binding. The binding was observed on α4β7-expressing transient and stable transfectants, and primary CD8+and CD4+ T cells. Of note, we showed that blockade or down-regulation of α4β7 expression on CD4+ T cells, the primary target of HIV-1 infectionin vivo, resulted in decreased HIV-1 infection.
α4β7 is not an indispensable receptor for HIV-1 entry (Arthos J, et al., 2008; Cicala C, et al., 2010; Cicala C, et al., 2009). We focused on investigating its role as an attachment factor. In agreement with previous reports (Arthos J, et al., 2008; Nawaz F, et al., 2011), we observed thatin vitrocellular expression of human integrin α4β7 conferred the capability for HIV-1 binding. Such binding likely increases the chance of HIV-1 binding to target cells. Our mutagenesis and pseudotyped virus experiments confirm that the α4β7-interacting site is located on the V2 loop of the envelope glycoprotein (unpublished data) (Arthos J, et al., 2008; McLellan J S, et al., 2011). Recent electron cryotomography has shed lights on the three dimensional architecture of native Env spike (Liu J, et al., 2008), demonstrating that the V1/V2 domain is located on the top of Env spike. In addition, the recent low-resolution crystal structure of α4β7 suggested that the long and wide groove between α4 and β7 subunits likely serves as the binding site for its ligands, including mucosal vascular addressin cell adhesion molecule 1(MAdCAM-1), Act-1 and a small antagonist (Yu Y, et al., 2012). Based on this finding, the crystal structure of a modeled full-length V1V2 loop was shown to be complementary to the groove of α4β7, resembling the binding to its natural ligands (Spurrier B, et al., 2014). Although it remains to be investigated, HIV-1 may utilize the V1V2 domain, protruding farthest away from viral membrane and nearest to the target cell, to interact with α4β7 and therefore facilitate viral attachment with the maximum steric advantage.
Of interest, we found that human α4β1 also exhibited the capability of mediating HIV-1 binding, albeit to a lesser extent. This is likely explained by that both α4β7 and α4β1 belong to the α4 integrin family and share common natural ligands including vascular cell adhesion molecule-1 (VCAM-1) and fibronectin, suggesting that structural similarity exists between them. Nevertheless, we observed that down-regulation of β1 did not affect HIV-1 infection. This was likely due to a low level expression of α4β1 on CD4+ T cells, and the observed marginal HIV-1 binding mediated by α4β1 was unlikely to be sufficient to enhance viral infection. Given that α4β1 is broadly distributed on many connective tissues in addition to lymphoid cells (Abram C L, et al., 2009; Yu Y, et al., 2012), targeting α4β1 likely affected HIV-1 infection by changing cell-cell interactions. However, the physiological implications of interaction between gp120 and α4β1warrant further study.
By using anti-α4β7 antibodies, we observed a decreased HIV-1 infection, demonstrating the potential significance of α4β7. It is noteworthy that the presence of anti-α4β7 antibodies could induce an obvious cell aggregation which varied from different antibodies used (Andrew D P, et al., 1994; Ruegg C, et al., 1992; Zeller Y, et al., 2001). Cell aggregation induced by α4β7 antibodies promotes cellcell contact, consequently facilitating cell-to-cell spread of HIV-1 infection. Therefore, α4β7 antibodies likely have counteracting effects on HIV-1 infection. Here the negative impact of α4β7 antibody blockade on virus binding to cells in suspension culture is likely offset by induced cellular aggregation, facilitating more efficient cell-to-cell spread of HIV-1. Whether virus infection was increased or decreased by antibody binding most likely depends on which one of the two effects is dominant. To avoid such confounders, we adopted shRNA-mediated interference to explore the impact of α4β7 down-regulation on HIV-1 infection. We demonstrated that transduction of α4-specific shRNA significantly decreased HIV-1 infection. As a control, and consistent with a previous report (Qin X F, et al., 2003), CCR5-specific shRNA almost completely suppressed virus infection, confirming the indispensable role of CCR5 and the dispensable role of α4β7 for HIV-1 infection.
Owing to the heterogeneity of HIV-1 strains, the conformation structure of Env trimer may vary markedly from strain to strain. It remains controversial that whether α4β7-utilization is a broad property across different HIV-1 strains, and the inconsistent observations from different groups may be attributed to differences in viral strains, cell targets, and different experiment designs (Arthos J, et al., 2008; Nawaz F, et al., 2011; Parrish N F, et al., 2012; Perez L G, et al., 2014). In addition, given the structure flexibility of integrin α4β7 and its multistep-regulated conformation (Luo B H, et al., 2007; Yu Y, et al., 2012), α4β7 utilization by HIV-1 in vivo may be very complex. Study of transmitted/founder (T/F) HIV-1 strains which are isolated at very early stage of HIV-1 infection is of particular importance to dissect the mechanism of HIV-1 mucosal transmission and establishment of early infection (Keele B F, et al., 2008; Kishko M, et al., 2011). It is shown that the envelope glycoproteins of early-transmitted strains feature compact variable loops and less N-linked glycosylation sites compared to strains isolated during chronic infection (Chohan B, et al., 2005; Derdeyn C A, et al., 2004; Sagar M, et al., 2006). Whether T/F strains can efficiently utilize α4β7 and identification of the sequence property of envelope proteins determining α4β7 utilization are the other two interesting topics which remain to be further explored. In conclusion, our current study suggests that α4β7 may serve as an attachment factor at least for some HIV-1 strains, providing a promising approach for investigating the potential roles of α4β7 in HIV-1 infection.
-
This work was supported by National Natural Science Foundation of China Grant 81273250 and Ministry of Science and Technology of China Grants 2013ZX10001005–003–002 and 2012ZX10001006–002.
-
The authors declare that they have no conflicts of interest. All human blood samples were collected under protocols approved by the Local Research Ethics Committee.
-
CL and QH conceived and designed the experiments. CL and JW performed the experiments. CL, RJS and QH analyzed the data. BW, TD, YL and RJS contributed to discussion. CL and QH wrote the paper.


 DownLoad:
DownLoad: