












HTML
-
Toll-like receptors(TLRs)are the most well characterized innate receptors that can trigger adaptive immuneresponses, including CD4+ T helper cell 1(Th1), cytolytic CD8+ T cell, and IgG2 antibody responses (Iwasaki and Medzhitov, 2010). While Th1 and cytotoxic T lymphocytes(CTLs)play a critical role in anti-viral defense, the molecular mechanisms through which TLRs preferentially trigger Th1 CD4+ and CD8+ T cell responsesremain to be fully understood. Naïve CD4+ T cells may differentiate into several T helper cell lineages, includingTh1, Th2, Th17, and regulatory T cells. T cell differentiation is influenced by the cytokine milieu and co-stimulatory signals provided by antigen-presenting cells, such asdendritic cells(DCs) and macrophages (Trinchieri, 2003; Trinchieri and Sher, 2007). Interleukin 12(IL-12)is theprincipal cytokine that directs Th1 polarization; however, TLR signaling alone induces only low amounts of IL-12 (Napolitani et al., 2005; Warger et al., 2006; Lyakh et al., 2008). Conversely, IL-10, which can be induced by TLRsignaling following infections and vaccination, plays an inhibitory role in Th1 CD4+ T cell differentiation (Moore et al., 2001; Saraiva and O'Garra, 2010). Therefore, characterization of the regulatory mechanisms underlyingTLR-induced IL-12 and IL-10 expression is highly relevant to a better understanding of host defense and vaccine development.
Recent studies have uncovered complex regulatory mechanisms involved in TLR-induced IL-12 and IL-10 production. IKKβ and c-Jun N-terminal kinase (JNK), which activate transcription factors nuclear factor κB (NFκB) and c-Jun, respectively, play an essential role in the induction of IL-12 through TLR signaling (Trinchieri, 2003; Lyakh et al., 2008; Zhu et al., 2008). In contrast, phosphoinositide 3-kinase (PI3K), tumor progression locus-2 (TPL2), extracellular signal-regulated kinase (ERK), and mammalian target of rapamycin (mTOR) have been implicated in the negative regulation of IL-12 expression in DCs and macrophages (Dillon et al., 2004; Ohtani et al., 2008; Schmitz et al., 2008; Weichhart et al., 2008; Kaiser et al., 2009; Aksoy et al., 2012; Pan et al.). It has been demonstrated that the TPL2-ERK pathway is critically involvedin the regulation of IL-12, which most likely occurs through the transcription factor, c-fos (Dillon et al., 2004; Kaiser et al., 2009). mTOR has recently been demonstrated to play a pivotal role in IL-12 expression, based on observations that rapamycin treatment led to elevated IL-12 expression in various innate immune cells (Ohtani et al., 2008; Weichhart et al., 2008; Haidinger et al., 2010; Wang et al., 2011). mTOR can form two distinct complexes, mTORC1 and mTORC2, but rapamycin mainly targets mTORC1, which is responsible for the phosphorylation of translation regulators S6 and 4E-BP1/2 (Huang and Manning, 2008; Laplante and Sabatini, 2012). AlthoughNFκB has been suggested to play a role in rapamycin-regulatedIL-12 expression (Weichhart et al., 2008; Wang et al., 2011), whether Rapamycin can directly regulate NFκBactivation remains controversial (Schmitz et al., 2008; Pan et al., 2012). Therefore, the key molecule through which mTORC1 regulates IL-12 expression remains unclear. On the other hand, TPL2-ERK, PI3K, and mTOR, along with p38 and IKKβ, constitutes a core kinase network to coordinate IL-10 expression (Saraiva and O'Garra, 2010). Nevertheless, the precise mechanisms by which TLRs regulateIL-10 expression remain to be identified.
Infection by hepatitis B (HBV) and C (HCV) viruses is the leading cause of liver cirrhosis and hepatocellular carcinoma (Kundi, 2007; Torresi et al., 2011; Swadling et al., 2013). With the successful development of HBsAg VLP-based vaccines, new HBV infections in healthy population have been largely controlled (Pichichero, 2008). However, elderly people, and patients with renal dysfunction or diabetes, remain vulnerable to HBV infection. Recent studies demonstrated that addition of monophosphoryl lipid A (MPL) to the conventional HBV vaccine regimen can induce faster and greater serological responses in renal dysfunction patients; this suggests that a potent Th1 response is necessary to improve HBV vaccination (Thoelen et al., 1998; Tong et al., 2005). With3-4 million new infections every year, HCV presents an even-greater challenge as the search for an effective vaccine continues. Studies from HCV-infected patients suggest that T cell responses-especially Th1 and CD8 responses-are well-correlated with spontaneous clearance of HCV (Cooper et al., 1999; Cox et al., 2005).Therefore, identification of strategies to induce potent and long-lasting Th1 and CD8 responses is critical for developing HCV vaccines.
To better understand the mechanisms through which TLR signaling induces IL-12 and IL-10 expression, and to fnd an effective approach to boost T cell responses for vaccine development, we systemically screened protein kinases involved in TLR-induced IL-12 expression. In the present study, we identifed up to fve distinct signaling pathways involved in the negative regulation of IL-12 expression in DCs. Accordingly, blockade of PI3K, ERK or mTOR can elevate IL-12 while attenuate IL-10 expression through c-fos, contributing to improved TLRinduced Th1 responses in vivo. Furthermore, blockade of mTOR by rapamycin in prime-boost models can considerably improve TLR-induced Th1 and antibody responses to HBV and HCV vaccines, providing new insights for the rational design of future vaccines.
-
LysM-cre mice were purchased from The Jackson Laboratory(Bar Harbor, ME, USA) and c-fos floxed mice were described previously (Min et al., 2012). C57/B6 and BALB/c mice were obtained from Shanghai Laboratory Animal Center(Shanghai, China). These mice were bred and maintained in a pathogen-free animal facility at Institut Pasteur of Shanghai.
-
Bone marrow cells were isolated by flushing the femur and tibia bones of 6-8 week-old C57BL/6 mice with RPMI 1640 medium (Invitrogen, USA). Red blood cells were lysed using ACK lysis buffer(0.15 mol/LNH4Cl, 1 mmol/L KHCIO3, and 0.1 mol/L Na2EDTA, pH 7.2). Granulocyte-macrophage colony-stimulating factor(GM-CSF, 20 ng/mL; R & D systems)was used to generate bone marrow-derived dendritic cells (BMDCs) in RPMI 1640 medium containing 5% fetal bovine serum (FBS; HyClone), 2 mmol/L L-glutamine, and 200 μmol/L β-mercaptoethanol. Medium and GM-CSF were replaced every 2 days. On Day 6, non-adherent cells were collected, centrifuged, and then re-suspended in fresh medium containing GM-CSF 1 day before use. To generate bone marrow-derived macrophages (BMDMs), bone marrow cells were cultured in RPMI 1640 medium supplemented with 30% L929-conditioned medium (containing macrophage colony-stimulating factor [M-CSF]), as well as 10% FBS. On Day 4, non-adherent cells were removed and fresh RPMI with L929-conditoned medium was added. BMDMs were used on Days 7-10. For preparation of peritoneal macrophages, 2 mL 3% (w/v) Brewer thioglycollate medium (R & D Systems) was injected into the peritoneal cavity of mice. After 3 days, mice were euthanized and the peritoneal cavity was flushed twice with 5 mL phosphate-buffered saline (PBS). Collected peritoneal cells were seeded on 10-cm cell culture dishes with complete RPMI 1640 medium (10% FBS). Non-adherent cells were washed off and adherent cells were stimulated the following day
-
To prepare whole cell lysates, cells were resuspended in lysis buffer (50 mmol/L Tris, pH 7.4, 150 mmol/L NaCl, 1% Triton X-100, and 1 mmol/L EDTA, pH 8.0) supplemented with protease inhibitor complete mini (Roche) and 1 mmol/L PMSF, 1 mmol/L Na3VO4, 1 mmol/LNaF. After incubation on ice for 20 min, cell debris was cleared by centrifugation at 13, 000 rpm for 15 min. To make nuclear extracts, cells were resuspended in hypotonic buffer(10 mmol/L HEPES, pH 7.6, 1.5 mmol/LMgCl2, 10 mmol/L KCl, and 1 mmol/L EDTA, supplemented with protease inhibitor complete mini(Roche), 1mmol/L PMSF, 1 mmol/L NaF and 1 mmol/L Na3VO4), and gently homogenized by Douncer(10-15 strokes).Following centrifugation at 3, 000 rpm for 5 min, supernatant was removed and pellet was further lysed in high salt buffer(20 mmol/L HEPES, pH 7.6, 500 mmol/LNaCl, 1.5 mmol/L MgCl2, and 1 mmol/L EDTA, supplemented with protease inhibitors and phosphatase inhibitors as described above)to make crude nuclear fraction.Nuclear fractions were further centrifuged at 13, 000 rpmfor 15 min to remove debris.
-
For immunoprecipitation, whole cell lysates were incubated with 2-5 μg antibodies against NFκB p65 (sc-8008, Santa Cruz Biotechnology Inc.) overnight and followed by incubation with 10-20 μL Protein A sepharoseTM6MB beads (GE Healthcare Life Sciences) for 1 h at 4 ℃. The immunoprecipitates were washed three times with wash buffer (50 mmol/L Tris, pH 8.0, 300 mmol/L NaCl, 1% Triton X-100, and 1 mmol/L EDTA). Proteins were eluted with 2 × SDS loading buffer by boiling for 10 min. Protein samples were resolved by 10% SDSPAGE and transferred onto PVDF member, which then probed by antibodies. p-S6 (Ser235/236), p-4EBP (Ser65), p-mTOR (Ser2448), p-TSC2 (Thr1462), p-p70S6K (Thr389), p-p65 (Ser536), p-IRF3 (Ser396), p-STAT1 (Y701), p-STAT3 (Y705), p-IκBα, p-IKKα/β, p-p38 (Thr180/Tyr182), p-JNK (Thr183/Tyr185); c-myc (#9402), c-fos (#2250), S6 (#2317), p65 (#4764), IRF3 (#4302) were purchased from Cell Signaling Technology. p-ERK (SC-7383), c-Rel (SC-71), IκBα (SC-1643) and Tubulin (SC-9935) were from Santa Cruz Biotechnology. Lamin B1 (#332000) was from Invitrogen, GAPDH (#AP0063) was from Bioworld, and p50 (ab7971) was from Abcam.
-
BMDMs were treated by DMSO or LPS (100 nmol/L) for 1 h, and then cross-linked by 1% Formaldehyde at room temperature for 10 min. Cells were then lysed in SDS lysis buffer (1% SDS, 10 mmol/L EDTA, and 50 mmol/L Tris, pH 8.0) and sonicated until the chromatin was sheared to an average size between 200 and 500 bp. Sonicated lysates were centrifuged at 13, 000 rpm for 10 min to clear the debris and the supernatants were diluted 10-fold by ChIP dilution buffer (50 mmol/L Tris, pH 8.0, 150 mmol/L NaCl, 1% Triton X-100, and 1 mmol/ L EDTA) and incubated with anti-p65(sc-372X, Santa Cruz Biotechnology Inc.) at 4 ℃ overnight. After incubation with protein A/G agarose for 1 h, the immunoprecipitates were collected and washed extensively with a series of wash buffer. The DNA/protein complexes were dissociated by heating at 65 ℃ for 5 h. Following RNase A and protease K treatment, DNAs were isolated by phenol/chloroform extraction and precipitated by ethanol.Precipitated DNAs were dissolved in water and analyzed by real-time PCR. All the buffers and reagents were fromUpstate Biotechnology(Billerica, MA)using the chromatin immunoprecipitation(ChIP)assay kit. PCR primers:
IL-12a: Forward 5′-GACCTGTCCCGGGACAAGAGT-3′ and Reverse 5′-CGCTGACCTTGGGAGACACAT-3′;
IL-10: Forward 5′-AATTCCACATTGGCTGTCCAGC-3′ and Reverse 5′-AGGATTATGCAAACCGGATTG-3′.
-
RNAs were extracted from BMDMs, BMDCs or peritoneal macrophages (pMφ) using TRIZOL (Invitrogen) according to the manufacture's instruction. cDNA was reversely transcribed from 1 μg total RNA by SuperScriptTM Ⅲ Reverse Transcriptase (Invitrogen). Real-time PCR were carried out with PrimeScript® RT reagent Kit (Takara) on ABI 7900HT Fast Real-time PCR System. Relative expression of target genes was quantitatively normalized against the expression of GAPDH using ΔΔct method.
-
Six-to-eight-week-old female BALB/c mice were intraperitoneally injected with 200 μL PBS, or PBS containing 10 μg ovalbumin antigen (OVA, InvivoGen), in various combinations of adjuvant lipopolysaccharide (LPS, 10 μg; InvivoGen), or LPS plus wortmannin (100 μg), U0126 (50 μg), rapamycin (100 μg), GO6983 (100 μg), or PP2 (30 μg). After 3 weeks, mice were re-stimulated with 1 μg OVA. One week after boosting with antigen, serum samples and spleen cells were collected, and 5 × 105 splenocytes/mL were stimulated with OVA-Ⅰ(10 μg/mL, SIINFEKL, InvivoGen)or OVA-Ⅱ(10 μg/mL, ISQAVHAAHAEINEAGR, InvivoGen) for 2 days.Supernatants were collected for evaluation of interferon gamma (IFN-γ) production (eBioscience) by ELISA, according to the manufacturer's instructions.
-
Six-to-eight-week-old female C57/B6 mice were intraperitoneally injected with 200 μL PBS, or 200 μL PBS containing 5 μg HBsAg (Shanghai YeaMen Biotech. Co., Ltd) with different adjuvants (10 μg MPLA [InvivoGen], 100 g rapamycin, 200 μg alum [InvivoGen], or MPL plus rapamycin or alum). After 4 weeks, mice were boosted once with 5 μg HBsAg and either MPL, rapamycin, alum, or MPL plus rapamycin or alum. Two weeks after boosting, serum samples and spleen cells were collected, and 5 × 105 splenocytes/mL were stimulated with HBsAg or CD8 peptide (ILSPFLPLL) for 2 days. Supernatants were then collected for evaluation of IFN-γ production by ELISA (eBioscience), according to the manufacturer'sinstructions, and 5 × 105 cells/well were plated and stimulated by HBsAg protein (10 μg/mL) or CD8 peptide (5 μg/mL ILSPFLPLL for 2 days). ELISPOT assays for IFN-γ and IL-4 were then conducted, according to the manufacturer's instructions. Antibody titers were measured by ELISA.
-
Soluble E2 protein from HCV 1b subtype isolate Con-1, containing a poly-histidine tag (His-tag), was produced in insect S2 cells and sequentially purifed using Nickel resin and HPLC. Twenty micrograms of E2 protein were intraperitoneally injected into 6-8-week-old female B6 mice, with or without adjuvants (10 μg MPLA, 100 μg rapamycin, 10 μg MPLA plus 100 μg rapamycin, 200 μg alum) in 200 μL PBS buffer. After 4 weeks, mice were boosted once with the same regimen. Six weeks after prime-boosting, mice received an E2 protein challenge (5 μg). One week after recall, spleen cells were collected. 1 × 106 splenocytes/mL were stimulated by E2 protein (10 μg/mL) for 2 days, and supernatants were then collected for IFN-production by ELISA (eBioscience), according to the manufacturer's instructions. Serum samples were collected 2 weeks after priming, 2 or 6 weeks after boosting, or 1 week after recall for IgG1 and IgG2b titers.
-
Two-tailed unpaired T test was used for statistical analysis and p < 0.05 is considered significant.*p < 0.05, **p < 0.01.
Mice
Generation of bone marrow-derived DCs and macrophages
Cell lysis and fractionation
Immunoprecipitation and Western blotting
Chromatin immunoprecipitation assay
Real-time PCR
Ovalbumin immunization and immune assays
HBsAg immunization and immune assay
HCV E2 immunization and immune assays
Statistical analysis
-
After viral infection, TLRs are the major innate receptors that trigger IL-12p70 production in DCs and macrophages. Comprehensive analysis of LPS-induced phosphorylation events suggests that in addition to IKKs and MAPKs, other protein kinases may also be involved in regulation of TLR-induced gene expression (Weintz et al., 2010). We therefore sought to determine the role of TLR-activated protein kinases in IL-12 induction and Th1 responses by screening a small library of kinase inhibitors.
By measuring the production of IL-12p70, a heterodimer composed of IL-12p40 and p35, we found that IL-12p70 production was reduced in LPS-treated BMDCsthat were pre-treated with various inhibitors(Figure 1A). Consistent with their well-established roles in TLRinduced IL-12 expression, inhibition of TAK1, IKKβ, JNK, GSK3-α/β, or p38 completely abolished IL-12p70 induction(Figure 1A). Unexpectedly, inhibition of seven other kinases, including p90RSK, casein kinase I, AMPK, calcium/calmodulin-dependent protein kinase Ⅱ (CaMKⅡ), ATM/ATR, polo-like kinase (PLK), and spleen tyrosine kinase (SYK) also impaired LPS-induced IL-12p70 expression(Figure 1A). These kinases have been previously implicated in various physiological processes, including DNA repair, metabolism and immune regulation, however, their roles in TLR signaling have not been established. Therefore, the potential involvement of these seven kinases in IL-12p70 expression underscores the complexity of LPS-induced signaling. Conversely, pre-treatment of BMDCs with various other inhibitors resulted in elevated IL-12p70 production(Figure 1B). These inhibitors target protein kinases belong to fve signaling pathways, namely mTOR-p70S6K, PI3K-Akt, Raf1-TPL2-ERK, Src and PKC pathway, respectively, and various inhibitors targeting different signaling components within the same axis of PI3K-Akt, TPL2-ERK and mTOR-p70S6K pathways resulted in similar effect on IL-12p70 production.

Figure 1. Identify protein kinases involved in LPS-induced IL-12p70 production and Th1 response. (A, B) BMDCs were pretreated by DMSO or indicated inhibitors for 30 min prior to the stimulation by LPS (100 ng/mL). Supernatants were collected in 24 hours and IL-12p70 was measured by ELISA. (C, D) 6-8 weeks old female BALB/c mice were intraperitoneally injected with 200 μl PBS, or PBS containing ovalbumin (10 μg) and LPS (10 μg), with or without wortmannin (100 μg), U0126 (50 μg), rapamycin (100 μg), GO6983 (100 μg), or PP2 (30 μg). In 3 weeks, mice were boosted once by OVA (1 μg). One week after boost, spleen cells (C) and sera samples (D) were collected. 5 × 105 splenocytes/mL were unstimulated or stimulated with OVA-Ⅰ (10 μg/mL, SIINFEKL) or OVA-Ⅱ (10 μg/mL, ISQAVHAAHAEINEAGR) for 2 days, supernatants were collected for ELISA measuring IFN-γ production. Data are presented as mean ± SEM of one representative experiment of at least three. Note: *p < 0.05, **p < 0.01, and N.D., Not detectable.
Subsequently, we focused on the top lead compounds targeting the aforementioned five negative regulatory pathways and assessed their effects on the Th1 response. Mice were immunized with OVA in the presence of LPS, with or without rapamycin(an mTOR inhibitor), wortmannin (a PI3K inhibitor), U0126(an ERK inhibitor), GO6983(a PKC inhibitor), or PP2(a Src inhibitor), and then challenged with OVA antigen after 3 weeks. One week after recall, spleen cells were isolated and stimulated by CD4 peptide OVA-Ⅱ and CD8 peptideOVA-Ⅰ ex vivo, and IFN-γ production was measured by ELISA. Although LPS induced strong OVA-specific T cell responses, rapamycin, wortmannin, and U0126, but not GO6983 or PP2, resulted in further elevation ofLPS-induced OVA-specific Th1 CD4+ and CD8+ T cell responses(Figure 1C). Moreover, rapamycin, wortmannin, and U0126, but not GO6983 or PP2, also augmented LPS-induced IgG2 subtype antibody production(Figure 1D). These results demonstrate that the protein kinases PI3K, ERK, and mTOR play critical roles in regulating the LPS-induced Th1 response in vivo.
-
To investigate why blockade of PKC and Src by GO6983 or PP2 failed to elevate the LPS-induced Th1 response in vivo, we examined the effects of these inhibitors on IL-12 and IL-10 induction 12 h after immunization. While rapamycin, wortmannin and U0126 greatly elevated LPS-induced IL-12 production in vivo, GO6983 and PP2 only modestly increased IL-12 production(Figure 2A). Moreover, unlike rapamycin, wortmannin and U0126, which resulted in considerable attenuation of IL-10 production, GO6983 and PP2 did not have any impact on LPS-induced IL-10 production in vivo(Figure 2A). These results indicate that the differential regulation of LPS-induced IL-12 and IL-10 expression by rapamycin, wortmannin and U0126 might have contributed to their augmentation of LPS-induced Th1 response in vivo.
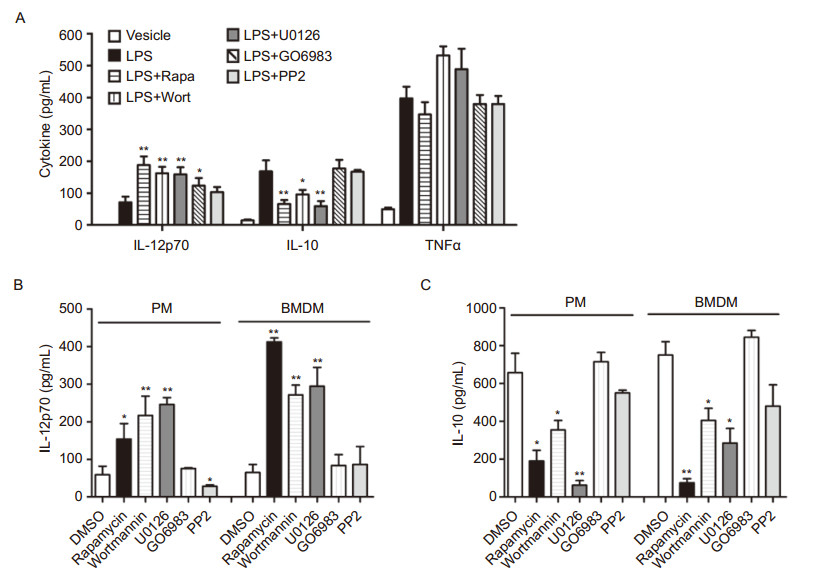
Figure 2. Investigate the role of protein kinases in LPS-induced IL-12p70 and IL-10 production in vivo and in vivo.(A)BALB/c mice(five per group)were immunized via I.P. with OVA(5 μg) and LPS(10 μg), with or without wortmannin(100 μg), U0126(50 μg), rapamycin(100 μg), GO6983(100 μg), or PP2(30 μg). In 6 hours, sera were collected from the tail veins of immunized mice. Secreted cytokines in sera were measured by ELISA.(B, C)Peritoneal macrophages and BMDMs were pretreated by DMSO or indicated inhibitors for 30 min prior to LPS stimulation for 24 hours. Supernatants were collected and IL-12p70 and IL-10 were measured by ELISA. Data are presented as mean ± SEM of one representative experiment of at least three. Note: *p < 0.05, **p < 0.01.
In addition to DCs, macrophages also produce IL-12p70, contributing to the induction of Th1 response (Trinchieri, 2003). Indeed, the mTOR inhibitor rapamycin, the PI3K inhibitor wortmannin, as well as the ERK inhibitor U0126, also signifcantly increased LPSinduced IL-12p70 production in peritoneal macrophages and BMDMs(Figure 2B). In contrast, the PKC inhibitor GO6983 only elevated IL-12p70 production in peritoneal macrophages, whereas the Src inhibitor PP2 failed to elevate IL-12p70 production in both macrophages(Figure 2B). Furthermore, rapamycin, wortmannin, and U0126 consistently decreased LPS-induced IL-10 production in macrophages, whereas PP2 only had a moderate impact and GO6983 had no impact on IL-10 expression (Figure 2C). Taken together, these data indicate that mTOR, PI3K, and ERK play critical roles in the regulation of IL-12 and IL-10 expression in the innate immune system, thereby modulating the LPS-induced Th1 response in vivo.
-
The results described above indicate that rapamycin, wortmannin, and U0126 exert similar effects on the expression of IL-12/IL-10 in DCs and macrophages. In the next part of the study, we examined the effects of mTOR, PI3K, and ERK on LPS-induced signaling pathways. Using Western blotting, we found that inhibition of PI3K, ERK, and mTOR did not alter the phosphorylation of IKKα/β, or its substrate, IκBα, in BMDMs; thus, these compounds did not directly affect IKKα/β or NFκB activation (Figure 3A). Conversely, we found that inhibition of PI3K, ERK, and mTOR decreased the phosphorylation of S6 and 4E-BP1/2, two substrates of mTOR(Figure 3A). While rapamycin did not affect the phosphorylation of tuberous sclerosis complex-2(TSC2), wortmann in and U0126 impaired the phosphorylation of TSC2 on residues Thr1462 and Ser664, respectively(Figure 3A).These results suggest that wortmannin and U0126 mainly impinge on TSC2, in order to inhibit mTOR activation.

Figure 3. Protein kinases PI3K, ERK and mTOR regulates LPS-induced IL-12 and IL-10 expression by c-fos. (A, B) BMDMs were either untreated or pretreated by U0126 (5 μmol/L), wortmannin (200 nmol/L) or rapamycin (100 nmol/L) followed by LPS (100 ng/mL) stimulation for various times. Cell lysates were prepared and resolved by 10% SDS-PAGE. Proteins were then transferred to PVDF membrane and immune blotted by respective antibodies. (C) BMDMs derived from c-fos floxed (WT) and LysM-cre:c-fos floxed mice (c-fos-/-)were left untreated or treated by LPS (100 ng/mL) for 4 hours. RNAs were prepared by Trizol and gene expression was measured by real-time PCR. (D, E) BMDMs derived from c-fos floxed (WT) and LysM-cre:c-fos floxed mice(c-fos-/-) were either untreated or pretreated by U0126 (5 μmol/L), wortmannin (200 nmol/L) or rapamycin (100 nmol/L), and followed by LPS (100 ng/mL) stimulation for 24 hours. Supernatants were collected for ELISA. These experiments have been repeated twice, and similar results were obtained. Data are presented as mean ± SEM, *p < 0.05, **p < 0.01.
As the TPL2-ERK pathway has been implicated in c-fos induction, we examined whether rapamycin, wortmannin and U0126 can also affect LPS-induced c-fos expression. Following LPS stimulation, c-fos was rapidly induced, reaching peak levels after 2 hours, but relapsing afterwards (Figure 3B). Interestingly, LPS-induced c-fos expression was impaired by rapamycin, U0126, and wortmannin treatment(Figure 3B), suggesting that c-fos may be a common effector of mTOR, PI-3K and ERK. To extend this finding, we generated wild-type and c-fos-/- BMDMs from c-fos floxed mice (defcient of c-fos in macrophages and neutrophils), respectively. After stimulation with LPS, we found that expression of IL-12a and IL-12b mRNA was elevated, while expression of IL-10 mRNA was diminished in c-fos-/- BMDMs(Figure 3C). Consistent with these results, higher levels of IL-12p70, but lower levels of IL-10, were secreted by c-fos-/- BMDMs after LPSstimulation(Figure 3D). More importantly, rapamycin, wortmannin, and U0126 were no longer able to increase IL-12p70 or to decrease IL-10 production in c-fos-/- cells(Figure 3E). These results demonstrate that c-fos plays a crucial role in mediating mTOR-regulated IL-12 and IL-10 expression in TLR4 signaling.
-
To investigate how c-fos regulates IL-12 and IL-10 expression, the promoter regions of IL-12a and IL-10 genes were cloned to pGL3-luc, generating plasmids pGL3-p12a-luc and pGL3-pIL-10-luc, respectively. Overexpression of NFκB p65, but not c-fos, promoted the expression of luciferase driven by pIL-12a and pIL-10 in HEK293 cells(Figure 4A, 4B). Interestingly, c-fos was able to diminish NFκB-mediated pIL-12a-luc expression, whereas to enhance NFκB-mediated pIL-10-luc expression in a dose-dependent manner(Figure 4A, 4B). By co-immunoprecipitation, we found that c-fos formed complexes with p65 in BMDMs stimulated by LPS(Figure 4C). Subsequently, chromatin immunoprecipitation (ChIP) assay was conducted to investigate the binding of NFκB to IL-12a and IL-10 promoters. In wild-type BMDMs, NFκB was efficiently recruited to promoters of IL-12a and IL-10 after LPS stimulation(Figure 4D). Compared to wild-type BMDMs, more NFκB binding was detected on IL-12a promoter, whereas less NFκB binding was present on IL-10 promoter in c-fos-/- BMDMs(Figure 4D). Taken together, these results suggest that c-fos may be able to modulate the DNA binding ability of NFκB in a promoter-specifc manner, thereby differentially regulate TLR-induced IL-12 and IL-10 expression.

Figure 4. c-fos regulates NFκB binding to the promoters of IL-12 and IL-10. (A, B) pGL3-luc containing 3.5 kb of IL-12a promoter (from -3363 to +127) (A) or 3.8 kb of IL-10 promoter (from-3770 to +69) (B) and plasmid expressing renilla luciferase were co-transfected with plasmid expression c-fos (Addgen, #19259), or/and plasmid expressing NFκBp65 (pcDNA3.0 vector) into HEK293T cells by calcium-phosphate method. Transfected cells were harvested 36 hours later and luciferase activities were measured. The data was presented as the relative frefly luciferase/ renilla luciferase ratio. (C) BMDMs were either untreated or treated by LPS (100 ng/mL) for 1 hour. Cell lysates were immunoprecipitated by anti-IgG or anti-p65, respectively. Immunoprecipitates were probed by anti-c-fos and anti-p65 sequentially. (D) BMDMs from c-fos floxed mice (WT) and LysM-cre:c-fos floxed mice(c-fos-/-)were either untreated or treated by LPS (100 ng/mL) for 1 hour. After cross-linking, cells were lysed and lysates were immunoprecipitated by anti-IgG or anti-p65, respectively. Immunoprecipitated DNAs were extracted and analyzed by quantitative real-time PCR. These experiments have been repeated twice, and the representative data are shown. Data are presented as mean ± SEM, *p < 0.05.
-
Since PI3K and ERK work in concert to activate mTOR, we then systemically analyzed mTOR-regulated innate immune gene expression induced by TLR signaling. In LPS-stimulated BMDMs, blockade of mTOR by rapamycin elevated the expression of IL-12a, IL-12b, IL-18, thymic stromal lymphopoietin (TSLP), and IL-23 mRNAs, whereas attenuated the expression of IL-10 mRNA(Figure 5A, 5B). Moreover, rapamycin treatment also increased the expression of co-stimulatory molecules CD80 and OX40L, but decreased the expression of inhibitory molecules PD-L1(Figure 5C, 5D).Conversely, rapamycin had little impact on a number of proinflammatory cytokines, including TNF-α, CXCL1, and IL-6, as well as IFN-β(Figure 5C, 5D). These results indicate that mTOR is involved in the regulation of a set of genes which are known to instruct the adaptive immune response. Similar to rapamycin treatment, c-fos defciency also resulted in increased IL-12a and IL-12b expression, as well as diminished IL-10 expression(Figure 3C). However, in contrast to rapamycin treatment, c-fos defciency did not alter the expression of TSLP or OX40L(Figure 5E). Moreover, while rapamycin did not affect IFN-β expression(Figure 5D), c-fos deficiency resulted in decreased IFN-β expression in BMDMs(Figure 5E). These results indicate that c-fos may be partially involved in mTOR-regulated gene expression induced by TLR signaling.

Figure 5. Rapamycin selectively regulates a set of innate genes involved in LPS-induced adaptive immune response. (A-D) BMDMs were pretreated by DMSO or rapamycin (100 nmol/L) for 30 min, and then stimulated by LPS (100 ng/mL) for 4 hours. (E) WT and c-fos-/- BMDMs were stimulated by LPS for 4 hours. RNAs were prepared and cDNAs were reversely transcribed for quantitatively analysis of gene expression by real-time PCR. Relative expression of target genes was quantitatively normalized against the expression of Gapdh. Data are presented as mean ± SEM of one representative experiment of three. Data are presented as mean ± SEM, *p < 0.05.
We subsequently investigated whether mTOR in fluencesthe activation or expression of transcription factors other than c-fos. In LPS-stimulated BMDMs, blockade of mTOR by rapamycin did not alter the phosphorylation of p65, or nuclear translocation of NFκB p65, p50, or c-Rel(Figure 6A, 6B). Moreover, rapamycin treatment did not affect the phosphorylation of the transcription factor IRF3, or MAP kinases JNK1/2 or ERK1/2(Figure 6A). In contrast, rapamycin impaired the phosphorylation of STAT3, and the expression of the transcription factors c-myc, hypoxia-inducible factor 1-alpha (HIF-1α), and C/EBPδ (Figure 6C), which may play roles in LPSinduced gene expression through feed-back mechanisms. Taken together, these results indicate that blockade of mTOR by rapamycin impairs the expression of several transcription factors, which may work in concert with c-fos to modulate the TLR-induced adaptive immune response.
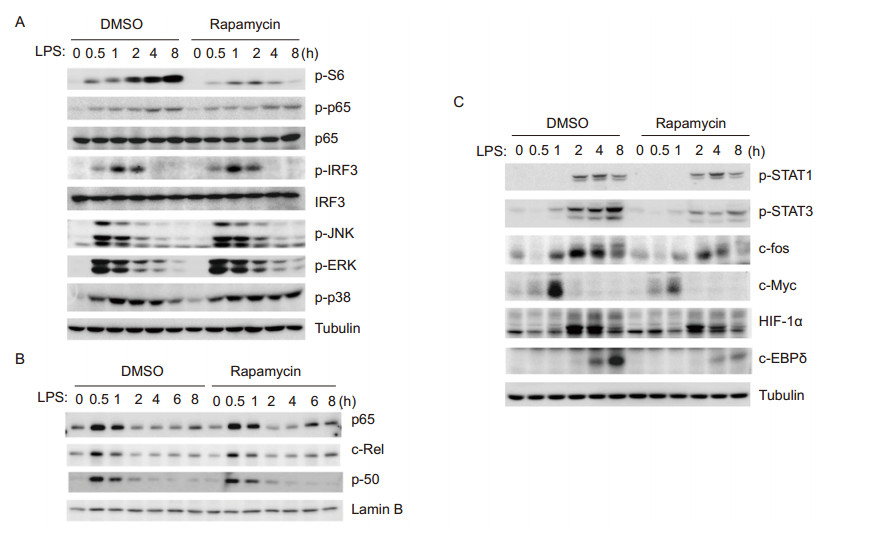
Figure 6. Blockade of mTOR by rapamycin impairs the expression of c-myc, HIF-1α and c/EBPδ, but not NFκB or MAPKs, in TLR4 signaling. BMDMs were pretreated by DMSO or rapamycin(100 nmol/L)for 30 min, followed by LPS(100 ng/mL)stimulation for various times as indicated. Whole cell lysates(A, C)or nuclear extracts(B)were prepared and resolved by 10% SDS-PAGE, and transferred blots were probed by antibodies as indicated. The experiments were repeated at least twice with similar results.
-
Next, we investigated whether blockade of mTOR by rapamycin can be a viable strategy to boost vaccine efficacy. The HBV vaccine composed of the virus-like particles (VLP) of HBV surface antigen (HBsAg) is one of the few successful recombinant vaccines in clinic use (Pichichero, 2008). However, the conventional HBV vaccine regimen using alum as adjuvant (HBsAg VLP + alum) achieved only limited efficacy in elderly population and renal-dysfunction patients. Interestingly, addition of MPL (a safe TLR4 ligand for human use) to the conventional HBV vaccine has demonstrated improved effcacy in renal-dysfunction patients (Thoelen et al., 1998; Tong et al., 2005), suggesting that better Th1 response is desirable for HBV vaccine. In this regard, we frst examined the effect of rapamycin on IL-12 production induced by a diversity of TLR ligands. Consistent with its effect on LPS, rapamycin efficiently increased IL-12 production in BMDCs stimulated by MPL, R848 and poly (I:C) (Figure 7A). Accordingly, rapamycin also augmented MPL-induced HBV-specific IFN-secreting CD4+ and CD8+ T cell responses in a dose-dependent manner, indicating that rapamycin can work together with MPL to improve vaccine effcacy (Figure 7B).

Figure 7. Transient Blockade of mTOR by Rapamycin elevates MPL-induced IL-12p70 and Th1 response for HBV vaccine. (A) BMDCs were pretreated by DMSO or rapamycin (100 nmol/L) for 30 min, and then stimulated by LPS (100 nmol/L), MPL (1 μg/mL), R848 (1 μg/mL) and poly (I:C) (20 μg/mL) overnight. The supernatants were collected and IL-12p70 production was measured by ELISA. Data are presented as mean ± SEM and *p < 0.05, **p < 0.01. (B) Mice (five per group) were I.P. injected with HBsAg VLP (5 μg) and MPL (10 μg), and various amounts of rapamycin as indicated, and boosted once by the same regime in 4 weeks. In two weeks, spleen cells were isolated and seeded (5 × 105 cells/well) on 96-well plates, and stimulated by HBsAg (10 μg/mL) or CD8 peptide (5 μg/mL, ILSPFLPLL) for 2 days. Supernatants were collected for the assessment of IFN-γ production by ELISA. Data are presented as mean ± SEM from 5 mice per group, and representative data from two independent experiments is shown. (C-E) Mice (five per group) were I.P. injected with HBsAg VLP (5 μg) in different combinations of adjuvant (rapamycin (100 μg), alum (200 μg), MPL (10 μg) + alum (200 μg) or MPL (10 μg) + rapamycin (100 μg)), and boosted once by the same regime in 4 weeks. In 2 weeks, spleen cells were isolated and seeded (5 × 105 cells/well) on pre-coated PVDF-membrane on microplates. After stimulation by HBsAg (10 μg/mL) or CD8 peptide (5 μg/mL) for 2 days, the positive spots were counted (C, D). Serum samples were collected for antibody assay by ELISA (E). Data are presented as mean ± SEM from 5 mice per group, and representative data from three independent experiments is shown. *p < 0.05, **p < 0.01.
We then compared the effect of rapamycin + MPL with two current adjuvant regimens, namely alum alone and alum + MPL, by a homologous prime-boost approach. One month after prime immunization by HBsAg with various adjuvants, mice were homologous boosted once (with the same antigen/adjuvant combination as the prime). In two weeks after homologous prime-boost, spleen cells were harvested and stimulated by HBsAg or CD8 peptide ex vivo for 2 days, followed by measuring IFN-γ-secreting T cells through ELISPOT. Compared to alum or alum + MPL, rapamycin + MPL induced much greater differentiation of IFN-γ-secreting HBVspecific CD4+ and CD8+ T cell responses (Figure 7C). Moreover, rapamycin + MPL induced much more IL-4-secreting T cells than alum or alum + MPL (Figure 7D). Furthermore, rapamycin + MPL elicited higher HBsAgspecific IgG2b/2c titers than alum or alum + MPL (Figure 7E). It is important to note that rapamycin alone did not have a significant impact on HBsAg-induced T cell or antibody responses (Figure 7C-7E). These results collectively suggest that the combination of rapamycin and MPL works better than current adjuvant regimens (alum or alum + MPL) for HBV vaccine.
-
HCV vaccination remains a major challenge worldwide, partly because strategies to induce balanced Th1 and antibody responses are limited. As the envelope protein E2 of HCV contains multiple epitopes for both T and B cells (Qiu et al., 2008; Naarding et al., 2011), we used truncated soluble E2 as an antigen to test whether rapamycin + MPL can induce superior immune responses against HCV. Twenty micrograms of E2 protein were administered by intraperitoneal injection, with or without adjuvants, using a homologous prime-boost approach. Sera were collected at various time points of the primeboost regimen, and antibody titers were measured by ELISA. E2 protein barely induced a detectable antibody response without adjuvant, and rapamycin alone failed to signifcantly boost the antibody response (Figure 8A). While MPL and alum induced similar levels of IgG1 antibodies, MPL induced much stronger IgG2 than alum (Figure 8A). Moreover, rapamycin + MPL induced much higher IgG1 and IgG2b antibodies than MPL or alum (Figure 8A). Furthermore, the antibody response elicited by rapamycin + MPL declined much slower and remained very high even at 6 weeks after boosting (Figure 8A).
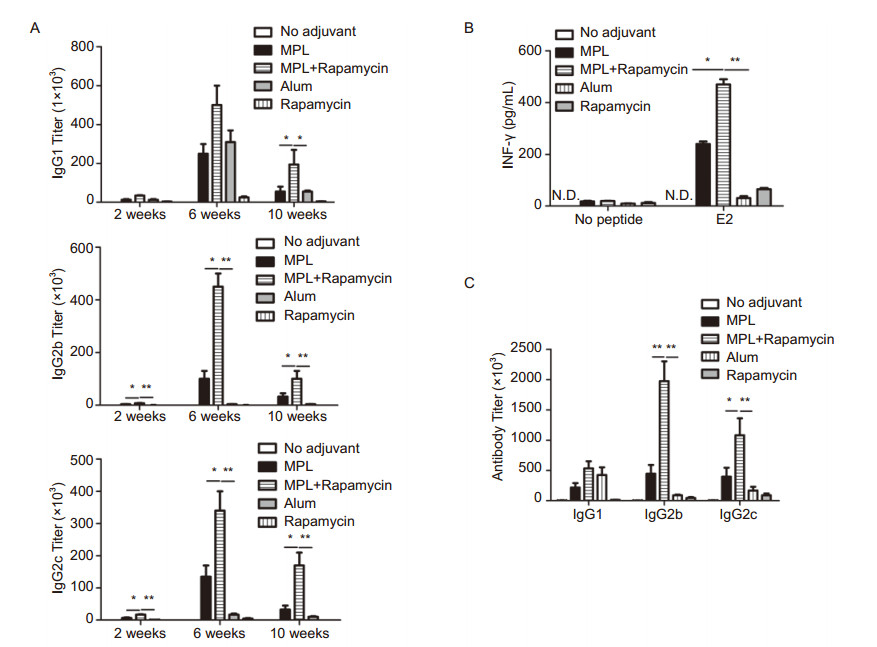
Figure 8. Transient blockade of mTOR by rapamycin improves MPL-induced Th1 and antibody response for HCV E2 subunit vaccines. (A) Mice (fve per group) were immunized via I.P. with soluble E2 protein (20 μg) alone, or with MPL (10 μg), rapamycin (100 μg), rapamycin (100 μg) + MPL (10 μg) or alum (200 μg), then boosted once with the same regimen in one month. Six weeks after boost, mice were challenged by 5 μg E2 protein. Sera samples were collected at various times after prime immunization and antibody titers were measured by ELISA. Note: 2 weeks, 2 weeks after prime; 6 weeks, 2 weeks after boost; 10 weeks, 6 weeks after boost. (B, C) one week after antigen challenge, spleen cells were harvested and stimulated (106splenocytes/mL) ex vivo by E2 protein (10 μg/mL) for 48 hours. Supernatants were collected and IFN-γ was measured by ELISA (B). Sera were collected and antibody titers were measured by ELISA (C). Data are presented as mean ± SEM from 5 mice per group, and representative data from two independent experiments is shown. Note: *p < 0.05, **p < 0.01.
Two months after prime-boost, the recalled immune response was analyzed. Compared with alum, MPL resulted in induction of more E2-specifc IFN-γ-producing T cells, indicating that MPL is a better inducer of the Th1 response to the HCV vaccine (Figure 8B). Moreover, rapamycin + MPL resulted in further elevation of the E2-specifc Th1 response, and induced more IFN-γ-secreting T cells than MPL (Figure 8B). In addition, rapamycin + MPL elicited the most potent antibody response upon recall (Figure 8C). Taken together, these results strongly suggest that rapamycin and MPL act in synergy to elicit a well-balanced and long-lasting adaptive immune response to the HCV vaccine.
Identifcation of protein kinases involved in TLR-regulated IL-12 expression and the Th1 response
Protein kinases PI3K, ERK, and mTOR are involved in LPS-induced IL-10 expression
Rapamycin, wortmannin, and U0126 regulate TLR-induced IL-12 and IL-10 expression by impinging on the mTOR pathway
c-fos regulates TLR-induced IL-12 and IL-10 expression through NFκB
mTOR regulates a special innate gene program involved in the TLR-induced adaptive immune response
Transient blockade of mTOR by rapamycin improves the TLR-induced Th1 response to the HBV vaccine
Rapamycin improves MPL-induced HCV vaccine effcacy
-
Although IL-12 plays a key role in the induction of the Th1 CD4 response, TLR signaling alone induces only low amounts of IL-12 in DCs and macrophages. TLR ligands -such as MPL and poly(I:C) -are being widely tested for vaccine use; thus, elucidation of the mechanism by which TLRs regulate IL-12 expression, and fnding strategies to boost the TLR-induced Th1 response, are highly relevant toward the rational design of vaccines against infectious agents and cancers. In this study, we took a systemic approach to identifying protein kinases critically involved in the regulation of TLR-induced IL-12 expression, and confirmed the role of a number of protein kinases -such as PI3K, ERK, mTOR, PKC, and Src -in regulating IL-12 expression in DCs and macrophages. Furthermore, we found that blockade of mTOR, PI3K, and ERK resulted in elevation of the Th1 response in vivo. Mechanistically, our study indicates that PI3K, ERK, and mTOR differentially regulate TLR-induced IL-12 and IL-10 expression, mainly through the transcription factor c-fos. Importantly, transient blockade of mTOR by rapamycin reprogrammed the TLR-induced innate gene program, enabling MPL to induce potent Th1 and antibody responses to HBV and HCV vaccines.
Although PI3K, ERK, and mTOR have been implicated in the regulation of IL-12 and IL-10 expression, the underlying mechanism has remained unclear. In this study, we first identified that the transcription factor, c-fos, acts as a common effector downstream of these kinases, mediating mTOR-regulated IL-12 and IL-10 expression in TLR signaling. It has been previously suggested that rapamycin may regulate LPS-induced IL-12 expression via NFκB. However, we and others failed to detect a direct effect of rapamycin on the activation and nuclear translocation of NFκB. Instead, we found that blockade of mTOR by rapamycin impaired the expression of c-fos, whereby modulating NFκB binding to IL-12a promoter. Therefore our study suggests a scenario that rapamycin elevates NFκB-mediated IL-12 expression by downregulating c-fos expression; this would reconcile disparate fndings of different studies. On the other hand, c-fos facilitated NFκB binding to the IL-10 promoter, which explains how rapamycin diminished IL-10 expression in TLR signaling by impairing c-fos expression. However, to determine how c-fos affects NFκB binding to different promoters in opposing ways, further investigations are warranted. While genetic ablation of TSC1 led to elevated JNK or ERK activation in LPSstimulated macrophages (Pan et al., 2012; Zhu et al., 2014), transient blockade of mTOR by rapamycin did not influence the activation of JNK, ERK, or p38. This suggests that mTOR may regulate MAPK activation through indirect, and as of yet, unknown mechanisms. Moreover, blockade of mTOR by rapamycin also affected the expression of HIF-1α, c-myc, and C/EBPδ, which may work in concert with c-fos to shape the TLR-induced innate gene program specialized in the regulation of the adaptive immune response.
So far, TLRs are the best characterized innate receptors that elicit the adaptive immune response. In addition to inducing DC maturation and promoting antigen presentation, TLRs also influence the differentiation of Th1 by cytokines IL-12 and IL-10. This study demonstrates that LPS-induced activation of PI3K, ERK, and mTOR in DCs and macrophages suppresses induction of the Th1 response, contributing to a better understanding of the underlying mechanism through which TLRs induce the T cell response. Importantly, transient blockade of mTOR by rapamycin can significantly improve HBV vaccine efficacy by acting in synergy with MPL in a prime-boost model. Compared with current HBV vaccine regimens, this provides a better strategy to elicit potent IFN-γ-secreting Th1 CD4+ and CD8+ T responses. In addition, our study also indicated that the combination of rapamycin and MPL can improve the magnitude and quality of the T cell response, for E2-based HCV vaccines. In addition to its effect on IL-12 and IL-10 expression, rapamycin augmented the expression of OX40L and TSLP, thereby facilitating Th2 differentiation and antibody responses, both for HBV and HCV vaccines. Thus, it is conceivable that a well-balanced T and B cell response, induced by rapamycin and MPL combined, would be highly benefcial in HBV and HCV vaccines. Furthermore, other effects of rapamycin on immune regulation also strongly support its potential for use in vaccines. For example, consecutive administration of rapamycin for up to 3 weeks after immunization can elevate the CD8 memory T cell response (Araki et al., 2009; Li et al., 2011; Amiel et al., 2012), and enhance Bacillus Calmette-Guérin (BCG) vaccine efficacy (Jagannath and Bakhru, 2012). In addition, DCs treated with LPS and rapamycin in vitro can result in improved anti-tumor immunity when transplanted into naïve recipient mice (Amiel et al., 2012). Therefore, investigations on different uses of rapamycin, as studied by our group and others, collectively indicate that rapamycin may be used to boost antigen-specifc immune responses of vaccines. Although rapamycin has been used in organ-transplanted patients and is widely considered to be an immune suppressive drug, we would point out that rapamycin has to act with other immune suppressive drugs -such as cyclosporine A -over a very long-term period, to have any immune suppressive effect (Almawi and Melemedjian, 2000). In contrast, in our approach, transient use of rapamycin along with MPL primarily enhanced antigen-specifc T cell responses.
In summary, this study not only reveals a new mechanism to better understand TLR-regulated IL-12 and IL-10 expression in the innate immune system, but also suggests a new strategy for boosting the TLR-induced Th1 response in vivo. As superior Th1 and CD8+ responses are highly favored in the control of infectious agents like HIV, HCV, and TB, as well as cancers, this study provides valuable insights relevant to vaccine development.
-
This study is support by National Natural Science Foundation of China grants 31070779 and 31170862 (to H.X.), 31100623 (to A.Z.) and 31270917 (to M.D.), and National Key laboratory of Virology grant (2014IOV006). H.X. is supported by Chinese Academy of Sciences "100-talent" program, National program for returned oversea talents, the CAS/SAFEA International Partnership Program for Creative Research Teams, and Shanghai Pasteur foundation.
-
The authors declare that they have no conflict of interest. All the animal procedures were conducted in compliance with the protocols approved by the Ethics Committee for Animal Care of Institut Pasteur of Shanghai, Shanghai, China, following the national guidelines and regulations for experimental animal use and welfare of China.
-
H.X. designed the research; L.H., A.Z., M.D., D.M., C.Y., C.Z., H.S., D.L. and X.H. performed the experiments. H.X., L.H., A.Z. and M.D. analyzed the data Q.D., J.X., K.L., S.X., H.Y., X.L., Z.H. and L.H. provided the reagents essential for the research. H.X. wrote the paper.


 DownLoad:
DownLoad: