













-
The chikungunya fever (CHIKF) is a severe disease with high incidence and wide prevalence in humans in tropical and subtropical countries and territories in the world. It is notably characterized by the sudden fever, severe arthralgia or arthritis in the extremities, and maculopapular rash on the truck and limbs (Laras et al., 2005). The etiological agent of CHIKF was named chikungunya virus (CHIKV) that belongs to the genus Alphavirus in family Togaviridae and was transmitted mainly by mosquitoes (Vanlandingham et al.; Diallo et al., 1999). CHIKV infection was often reported in Africa and Southeast Asia. The first CHIKV infection was identified in 1953 during a massive outbreak of febrile illness in Tanzania (Robinson, 1955). Since then, large outbreaks and epidemics of CHIKF were reported in Africa, Southeast Asian countries, and Indian subcontinent (AbuBakar et al., 2007). After a large outbreak in Indian in 1973, the virus activity virtually disappeared for almost 20 years that only sporadic CHIKF cases were recognized in Southeast Asian countries during the 1980s to 1990s (Lam et al., 2001; Arankalle et al., 2007). In 2004, CHIKV reemerged in Kenya and leaded to the widespread infection to Indian Ocean islands and Indian subcontinent where millions of people were infected (Arankalle et al., 2007; Casal et al., 2015). In 2007, CHIKV was imported to European countries (Italy and France) by viremic travelers, transmitted to local residents by mosquito bites, and thus caused autochthonous CHIKV cases (Rezza et al., 2007; Delisle et al., 2015). In 2013, autochthonous CHIKV cases on Saint Martin Island in Americas were confirmed. Soon, other territories including Caribbean, Central America, South America and North America also reported CHIKV infection (CDC, 2006; Leparc-Goffart et al., 2014).
The genotypes of CHIKV strains were suggested to be associated with spatial and temporal associations. Phylogenetic analysis has showed that CHIKV strains could be divided into three major genotypes, the West Africa (WA), Eastern/Central/Southern Africa (ECSA), and Asia lineages (Volk et al., 2010). Since the reemergence of CHIKV activities in 2000 in Southeast Asia, the newly discovered CHIKV strains have evolved to be distantly related to the old strains, which constituted two new sub-lineages. One sub-lineage designated as Indian Ocean lineage (IOL) evolved from ECSA, the members of which was circulating mainly in the Indian Ocean islands and Indian subcontinent and caused magnitude epidemics during 2005–2009. The other sub-lineage originated from Asia and was assigned as Asia.reemerge, which triggered large pandemics in the American countries and territories during 2014–2015 (Kariuki Njenga et al., 2008; Volk et al., 2010). So, the transmission of CHIKV among the tropical and subtropical countries still posed significant threat to the health of the local residents.
During 2016 to 2017, an outbreak of CHIKV infection of more than 3,000 cases occurred in Pakistan where large-scale CHIKV infection has never been reported before (Aamir et al., 2017). Ten samples of human sera were collected from clinical CHIKF patients in the Kalachi outbreak in Pakistan. Eight different strains were isolated from C6/36 cells incubated with the sera, and the genome of one strain (Pakistan-03, GenBank accession number: MF740874) has been completely sequenced by Dr. Liu et al. (2017) . In this study, we reported the whole genomic sequences of the other 7 CHIKV isolates. Comprehensive phylogenetic and evolution analyses were carried out including the sequences of the 8 Pakistani isolates and other strains deposited in GenBank so far. Our study reviewed the temporal and spatial history of CHIKV epidemics and outbreaks and conducted migration analyses of CHIKV strains. And the evolutionary statues and geographic origination of the Pakistani CHIKV isolates were investigated. These findings would be important for the better understanding of CHIKV evolution and transmission, which further facilitates the prevention and control of CHIKV infected disease.
-
Eight CHIKV isolates were obtained by blind passages from sera samples of eight patients with CHIKV infection in Pakistan in 2016 as described (Liu et al., 2017). Besides the complete genome of strain Pakistan-03 (GenBank no. MF740874), the complete genome sequences of other 7 strains were also obtained using the specific primers (Supplementary Table S1). The strains were named in accordance with the numbers of serum samples as Pakistan-01, Pakistan-04 to Pakistan-07, Pakistan-09, and Pakistan-10, and the genome sequences were deposited in GenBank to get access numbers (GenBank no.MF774613 to MF774619).
-
Besides the eight sequences from Pakistan, the CHIKV genomic sequences deposited in GenBank until 1st May 2017 were used for the dataset construction, including the corresponding information of collection dates and locations. In total, 309 sequences from 41 different countries were used. We constructed two datasets, the NSP309 dataset containing the non-structural protein (NSP) coding sequences and the SP309 containing the structural protein (SP) coding sequences. Sequences were aligned using ClustalW alignment tool within MEGA (Kumar et al., 2008).
-
The generalized time reversible plus gamma distribution (GTR+G) model was identified using jModelTest (Posada, 2008) as the best-fit model for phylogenetic analyses with both NSP309 and SP309 datasets. The Bayesian Markov chain Monte Carlo (MCMC) method available in the BEAST v2.3.0 package (Drummond and Rambaut, 2007) was used to estimate divergence times, substitution rate, and the phylogeographic analysis with the GTR+G4 model of nucleotide substitution, a strict clock model and coalescent constant population tree prior. For each dataset, BEAST analysis were run for 40 million generations to achieve convergence of parameters by calculating the effective sample size (ESS > 200) using TRACER version 1.6 (http://tree.bio.ed.ac.uk/software/tracer/). The maximum clade credibility (MCC) tree was computed using the TreeAnnotator program within the BEAST package with the first 10% trees removed as burn-in and visualized via Figtree v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/).
-
Alignments of NSP and SP sequences belonging to defined lineages were carried out respectively using MEGA. The non-consensus sites employing variable amino acids were noted to obtain the consensus sequences corresponding to each lineage. If more than 30% of the strains belonging to one lineage used an identical amino acid different from other sequences at the non-consensus site, it would be considered as choices of the consensus sequences at the sites for this lineage. Therefore, there usually will be one to three amino acids at those sites representing the consensus sequences of each lineage. The consensus sequences from each lineage were aligned to find the lineage-specific varieties according to the following principles: (a) a site employed one same amino acid for all strains from a same lineage but different from other lineages was considered to be the extra-lineage specific variation; (b) a site using two or three different amino acids within one lineage was considered to be the intra-lineage specific variation.
-
To better clarify the migration pathway of CHIKV among continents, the sequences from 41 different countries were catalogued to 12 geographic divisions (Supplementary Table S2). The eight isolates from Pakistan were retained as a separate geographical taxon so as to elucidate the geographic origins of these strains and epidemics from Pakistan. The dataset SP309 combined with the discrete variable of location information was subject to BEAST package using the same model as phylogenetic analysis for spatial-temporal dispersion pattern analysis. FigTree v1.4.3 program was used to generate MCC tree with the internal branches colored according the estimated origin. The transmission pathways among regions were illustrated in a world map.
-
The Bayesian phylogenetic trees were constructed based on the SP309 and NSP309 datasets respectively (data not shown), which had identical tree topology similar to the tree constructed with full-length open reading frames (ORFs) sequence as described (Volk et al., 2010). Then we used the phylogenetic tree based on the SP309 dataset for the subsequent analyses (Figure 1). As revealed in Figure 1, the epidemic CHIKV strains had an ancestor of about 1600 years ago, and now could be classified as three major lineages (West Africa [WA], East Central South Africa [ECSA], and Asia lineages) with close association with their geographic distributions and epidemic times. The lineage WA included all viruses from West Africa detected before 1993. Based on the phylogenetic Bayes tree, the most common ancestor (tMRCA) of the WA lineage could be dated to 1953 when the first CHIKV strain was isolated in Tanzania (Lumsden, 1955). The ECSA and Asia lineages evolved separately from WA. They shared a common ancestor until 1874 and then diverged from each other. The history of epidemics caused by strains of Asia lineage could be traced back to 1952, earlier than the Thailand outbreak in 1958 when the first CHIKV epidemic in Asia was confirmed (Volk et al., 2010). The Asia lineage was then divided into two sub-lineages with the tMRCA dated to 1996. The Asia.old sub-lineage mainly contained the strains identified in South East Asia during 1958–1996, and the Asia.reemerge sub-lineage comprised the strains responsible for epidemics in America and islands in Caribbean during 2014–2015, which indicated the introduction of CHIKV into to North/South America and Caribbean Sea islands from Southeast Asia. According to the spatial and temporal distribution of CHIKV strains, the ECSA lineage was diverged into two sub-lineages, the ECSA.Africa (old genotype) and the ECSA.IOL (new genotype). ECSA.Africa composed of strains from the epidemics in Eastern/Central/Southern Africa mainly from 1953 to 1984 and four isolates identified from 2014 to 2015 in Southern Africa (Figure 1), indicating the existence of the old genotype in this area after a disappearance of virus activity for more than twenty years. The ECSA.IOL sub-lineage mainly consisted of viruses responsible for the outbreaks in islands of Indian Ocean and Indian subcontinent region from 2005 to 2009, and outbreaks in Southeast Asia from 2007 to 2013. In addition, we noted a few strains belonging to the lineages inconsistent with their spatial and temporal distributions. The basal location of isolates from Kenya was observed in the ECSA.IOL sub-lineage (Figure 1), which supported the speculation that the 2004 epidemic in Kenya promoted the evolution of IOL strains and their subsequent migration to Indian Ocean islands and Indian subcontinent (Kariuki Njenga et al., 2008). One strain isolated from Indian 1986 was included in ECSA.Africa lineage, suggesting CHIKV spreading from Africa to Indian at least 20 years before the 2004 epidemic (Figure 1). Therefore, these discrepancies suggested migrations and genetic exchanges among different lineages.
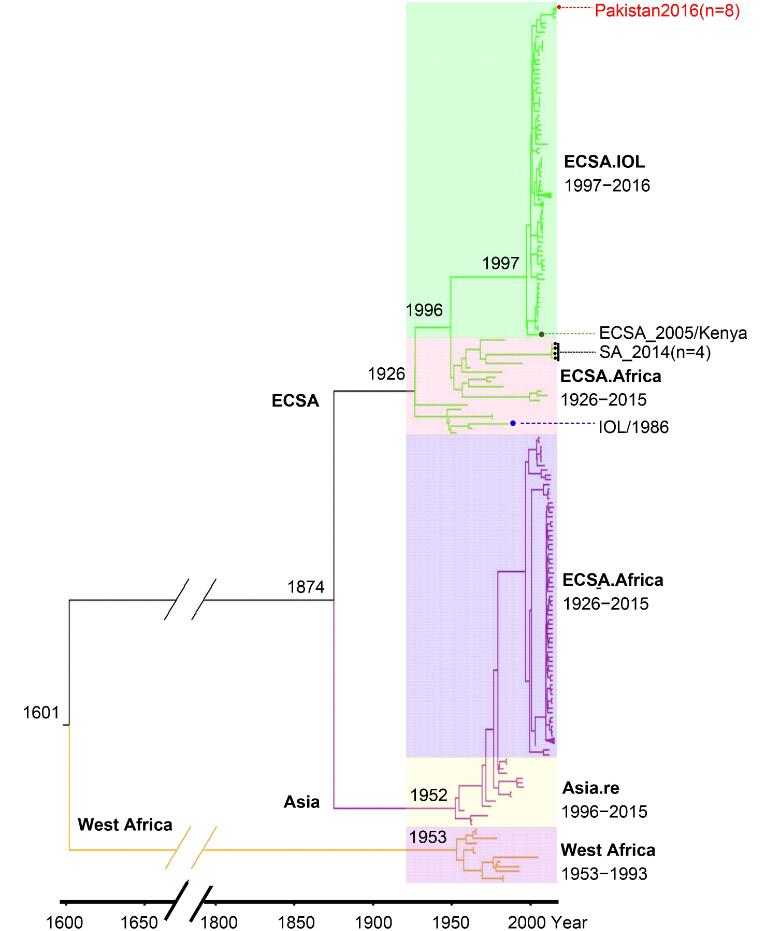
Figure 1. Evolution of 309 CHIKV strains basing on sequences coding structural proteins. The MCC Bayesian tree was based on strict clock model and coalescent constant population tree prior. The three major lineages are highlighted with different branch colors. And the sub-lineages are distinguished by translucent colored rectangle. The estimated time for the most recent common ancestors are labeled beside the node. Strains highlighted by red solid dot represented CHIKV isolated from Pakistan. One strain highlighted by green solid dot represents the speculated origination of IOL sublineage. Four strains high-lighted by black solid dot represent the recent CHIKV (isolated in 2014 or 2015) belonging to the ECSA. Africa sub-lineage. One strain highlighted by blue solid dot represents CHIKV isolated from Indian Ocean Islands but belongs to ECSA. Africa sub-lineage
It was the first time that CHIKV isolates were reported from Pakistan. Pairwise comparison of the nucleotide sequences of the Pakistani isolates showed that they shared very high similarities (99%) to each other except that the strains Pakistan-03 and Pakistan-04 shared 98% similarity. The eight strains from Pakistan were also involved in the phylogenetic tree, which showed that they clustered together and belonged to the ECSA.IOL sub-lineage. Phylogenetic analysis also showed that the Pakistan strains were mostly close to the strains identified from Indian in 2016 (Figure 1).
-
The CHIKV genomic sequence had two ORFs, one encoding a non-structural polyprotein precursor and the other encoding a structural protein precursor. The non-structural polyprotein precursor would be cleaved into four NSPs (nsP1, nsP2, nsP3 and nsP4) and played roles in viral RNA replication and translation. The structural protein precursor would be cleaved into five SPs: core protein (C), 6K/Transframe protein (6K), envelope proteins (E1, E2, and E3) and was suggested to be related to the host specificity of CHIKV. We investigated the lineage-specific amino acid varieties in both structural and non-structural proteins based on the 309 CHIKV sequences (Figure 2, Table 1). In total, we found that 32 sites had amino acid varieties in the Asia.reemerge sublineage, 13 in NSPs and 19 in SPs, 22 sites in the Asia.old lineage with 9 in NSPs and 13 in SPs, and 14 sites in the ECSA.IOL lineage with 8 in NSPs and 6 in SPs. Varieties were found in much fewer sites in the ECSA.Africa with 3 in NSPs and 2 in SPs, as well as the WA lineage with 2 in NSPs and 1 in SPs. We further calculated the evolutionary rate of each lineage based on the SP309 dataset according to the phylogenetic analyses (Table 2). We found that the ECSA.IOL sub-lineage had the highest substitution rate. Overall, the ECSA.IOL, Asia.reemerge and Asia.old lineages had higher evolutionary rates than the ECSA.Africa and WA lineages. So, the lineages with higher evolutionary rates might evolve more rapidly and resulted in varieties in more sites. We also found that among all lineages, E2 protein presented varieties in 11 sites and was the most variable protein comparing to other viral proteins. NS3 was the most stable protein with only a 4aa deletion in Asia.reemerge sub-lineage. The eight strains from Pakistan presented specific variations in a few sites comparing to other IOL strains. Most IOL strains employed L539 in NS2, R82 in NS4, E211 and A226 in E1, and K252 in E2. Differently, all 8 strains used S539 in NS2, S82 in NS4, K211 and V226 in E1, and Q252 in E2. The differences from the Pakistani strains might indicate the further evolution of the strains during the epidemics. Subsequently, to better understand the evolutionary influences on CHIKV genotypes, we characterized the detailed varieties in viral proteins of the two newly derived sub-lineages.
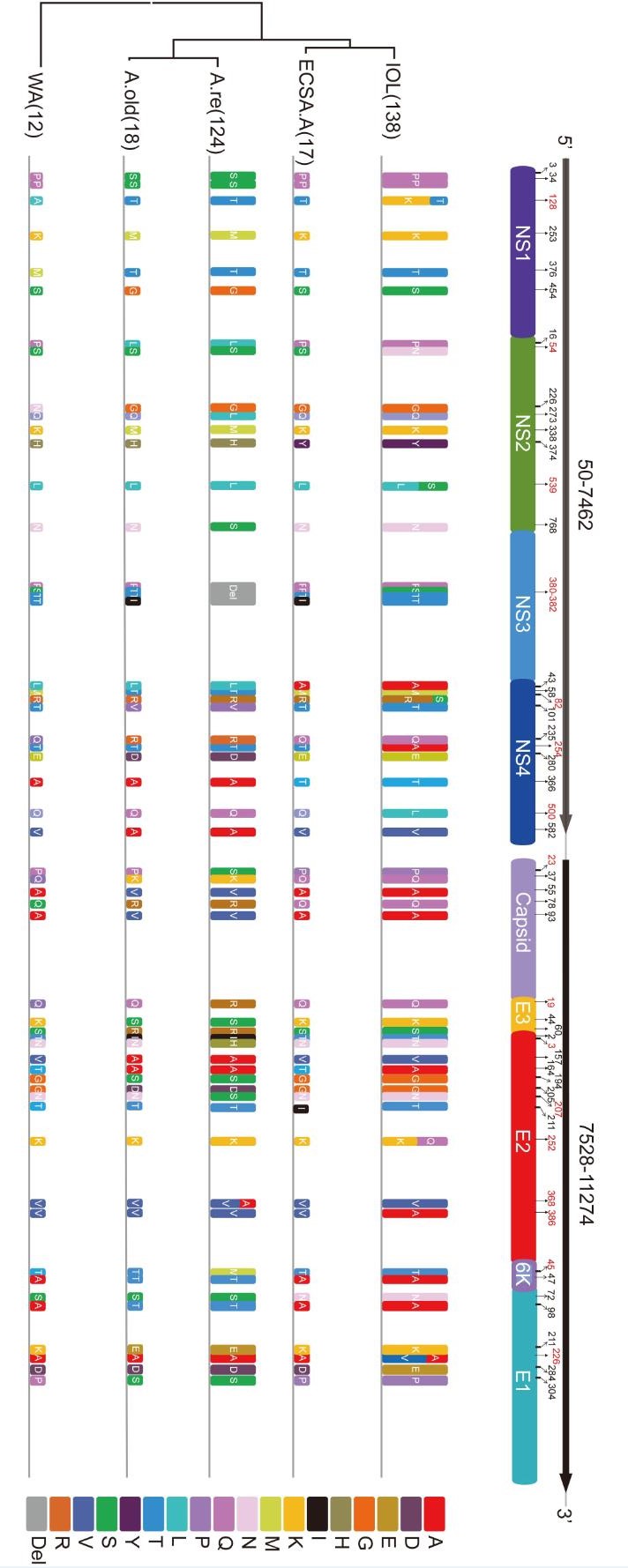
Figure 2. The Lineage-specific varieties in non-structural and structural proteins. The amino acid varieties in sites are represented by rectangles in different colors. The amino acid deletion is shown in grey box. The height of the rectangle is in proportion to the amount of stains in each lineage. The amino acid positions in each viral protein are indicated by corresponding numbers and those related to the lineage-specific varieties are labeled in red numbers.
Lineage NS1 NS2 NS3 NS4 C E3 E2 K E1 Asia.reemerge P/S3
K/M253
S/G454P/L16
Q/L273
K/M338
N/S768Del:380-382 M/T58
T/V101
Q/R235
E/D280
V/A582P/S23
Q/K37
A/V55
Q/R78
A/V93Q/R19
K/S44
S/R60T/I2
N/H3
V/A157
G/S194
G/D205
N/S207
V/A368T/M45
A/T47A/T98
P/S304Asia.old P/S3
K/M253
S/G454P/L16K/M338 M/T58
T/V101
E/D280
V/A582Q/K37
A/V55
Q/R78
A/V93K/S44
S/R60T/I2
V/A157
G/S194
G/D205A/T47 A/T98
P/S304ECSA.IOL T/K128 S/N54
H/Y374
L/S539
L/A43
R/S82
T/A254
A/T366
Q/L500K/Q252
V/A386S/A72
E/K211
A/V226
D/E284ECSA.Africa H/Y374 L/A43A/T366 A/T164 S/N72 WA T/A128 G/N226 A/T164 Table 1. Lineage-specific mutations of CHIKV. The different mutations in sites of CHIKV from Pakistan comparing to most other IOL strains were indicated in bold characters
Lineage Evolutionary rate
(subs/nt/year)Transmission
patternAsia.old 4.078E-4 epidemic Asia.reemerge 2.596E-4 epidemic ECSA.Africa 2.27E-4 enzootic ECSA.IOL 7.43E-4 epidemic West Africa 2.203E-4 enzootic Total 2.978E-4 -- Table 2. Evolutionary rate of each lineage
-
The ECSA.IOL specific varieties. All strains in ECSA.IOL lineage used A386 in E2 protein, which could be significantly distinguished from other lineage (Figure 2, Figure 3A, Table 1). However, this extra-lineage specific substitution was not described previously and the influence of the substitution remained to be investigated. Moreover, while strains of ECSA.Africa lineage all used A226 in E1 protein, the strains of ECSA.IOL lineage used either A or V at position 226 (Figure 2, Figure 3A, Table 1). It was suggested that the use of V226 in E1 protein made the ECSA.IOL strains more adaptive to the A. albopictus, which was more widely distributed than other related mosquito vectors in the world. Some IOL strains with V226 mutation in E1 protein further acquired a K to Q change at position 252 in E2 protein (Figure 2, Figure 3A, Table 1). This acquisition was reported to be related to enhanced infection efficiency to A. albopictus, which was experimentally confirmed using a reverse genetic system of CHIKV (Tsetsarkin et al., 2014). As with the strains from Pakistan, no the adaptive-related substitutions specific for the IOL lineages were obtained, as A226 in E1 protein and K252 in E2 protein were employed (Table 1). The IOL-specific varieties in non-structural proteins were found including extra-lineage variations in three sites that distinguished the IOL strains from the others (N54 in NS2, and A254 and L500 in NS4) and two sites within the IOL lineage (T/K128 in NS1, L/S539 in NS2 and R/S82 in NS4) (Figure 2, Table 1).

-
The Asia.reemerge lineage-specific substitutions. In the Asia.reemerge sub-lineage, we found a region of 4 aa deletions from positions 380 to 383 in NS3 protein among 107 isolates (Figure 3B). Besides, two strains from Malaysia and one from New Caledonia had a second deletion from positions 385 to 387 in NS3 (Figure 3B). We noted that the strains with the double deletions were identified in 2006 and 2011 respectively, which were early than those strains with one region of deletion. Since they located at a basal position within the Asia.reemerge sub-lineage, it is presumed that the CHIKV strains of Asia.reemerge sub-lineage might have the double deletions at the beginning of their emerging epidemics, subsequently experienced recombination with other strains lacking the second deletion in NS3 so as to acquire 4 amino acids at the positions.
In addition, the Asia.reemerge sub-lineage had a lineage-specific substitution at position 45 in 6K protein, which could be distinguished from other lineages significantly (Figure 2, Table 1). Within the sub-lineage, variations at four sites (N/H3, N/S207, V/A368) in E2 protein, one in E3 protein (Q/R19), one in Capsid protein (P/S23) were specific for the Asia.reemerge strains. In non-structural protein, the unique lineage-specific substitution at position 273 in NS2 protein (L273) distinguished the Asia.reemerge from others (Figure 2, Table 1). Since the epidemic of Asia lineage in South East Asia, it has been nearly ten years that the Asia.reemerge lineage with these unique substitutions emerged in Southeast Asia again and especially in the new territory including the Americas. So it is presumed that the emerging lineage might have acquired advantages from the genetic evolution for the better survival and adaptive abilities which results in a boarder range of epidemics in America. Such relationship needs further investigation and presently few studies were focused on the Asia.reemerge varieties analysis.
-
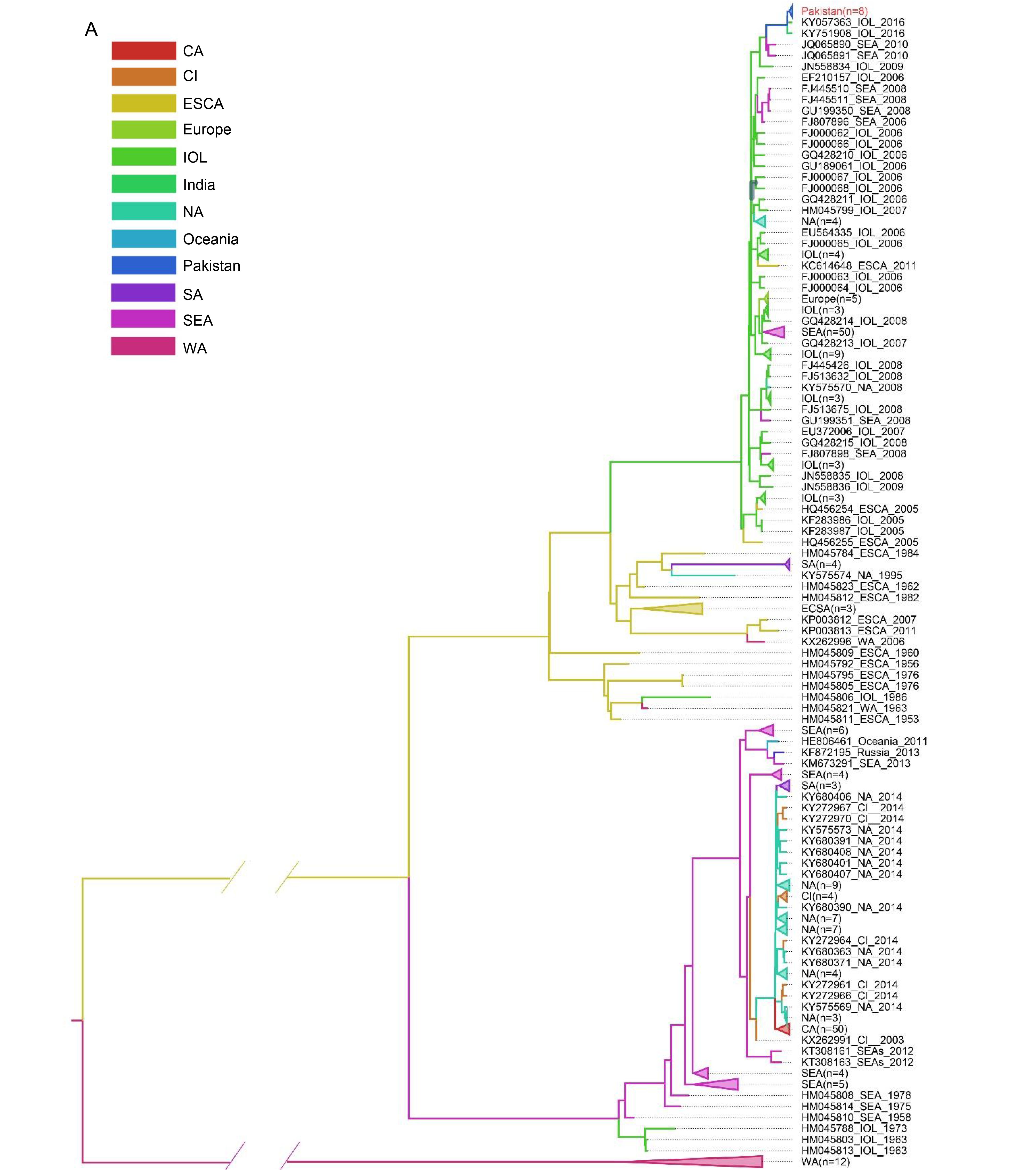
According to the phylogenetic analysis, the CHIKV strains could be divided into 3 major lineages originated from Africa and Asia. The 309 strains from 41 countries were catalogued as 12 regions were exhibited in the MCC tree with branches of corresponding colors (Figure 4A). The appearance of different colors under a major branch suggested the potential movement events among the different regions indicated by colors. Obviously, no migration events were observed in the West Africa lineage (Figure 4A), which was consistent with the results from phylogenetic analysis. To better understand the global movements of CHIKV strains, we mapped the spreading pathway among the 12 regions in a world map (Figure 4B). In general, CHIKV spreading mainly occurred in the Asia.reemerge and ECSA.IOL lineages. The Asia.reemerge was transmitted from Southeast Asia (mainly about Malaysia, Indonesia and Philippine) to India and Americas (including North America, Islands of Caribbean and Brazil) as well as Pacific Islands. Notably, French Caribbean island of Saint Martin was suggested to be the transmission origin of the epidemics in America, which was quite coherent to the fact that the first CHIKV in America was identified in Saint Martin in 2013 (Cauchemez, 2014). The spread pathway of ECSA.IOL mainly originated from Kenya. The viruses first moved to islands of Indian Ocean and Indian continent, and then spread to Southeast Asia. The transmission events from India to Europe (Italy and France) in 2006 were confirmed to be related with infected travelers (Parola, 2006). Finally, the newly emerging epidemics in Pakistan were suggested to be imported from Indian continent (Figure 4B).
-
Phylogenetic analyses suggested that the epidemic genotypes of CHIKV strains were tightly associated with the spatial and temporal distributions, which were named as the WA lineage mainly from Western African countries, the Aisa.old and Asia.reemerge lineages mainly from Asia, and the ECSA from the Eastern, Central, and Southern Africa. It has been suggested that the distribution of CHIKV outbreaks and epidemics were related to the local mosquito species (Weaver, 2013; Weaver and Forrester, 2015). Aedes spp. was considered to be the major vectors and was widely distributed in the tropical and subtropical countries (Ngoagouni et al., 2017). In Africa, CHIKV persisted in a sylvatic, enzootic cycle which involved much diverse non-human reservoir hosts, such as bats, and primatophilic mosquito species (A. furcifer-taylori, A. africanus, A. luteocephalus, and A. neoafricanus). In contrast, CHIKV was mainly associated with urban cycle transmitted by anthropophilic mosquito species (A. aegypti and A. albopictus) to human hosts in Asia (Diallo et al., 1999). So, the CHIKV from Africa and Asia were transmitted in different cycles, suggesting that the Asia and ECSA.IOL lineages might have evolution mechanisms different from the WA and ECSA.Africa lineage. Such differences were also reflected by the higher evolutionary rate of CHIKV involved in epidemic cycle (ECSA.IOL, Asia) and relatively lower evolutionary rate of CHIKV subjected to enzootic transmission cycle (WA, ECSA.Africa). So it is presumed that the more diversity of CHIKV infected hosts and vectors might constrain the evolution rate of ECSA.Africa and WA lineage related to enzootic cycle comparing with the Asia CHIKV strains in the epidemic cycle (Diallo et al., 1999).
The adaptive ability to the species of mosquito hosts might also affect the distribution and migration of CHIKV strains belonging to different lineages. We analyzed the specific genetic changes of CHIKV strains from different lineages. E1 and E2 proteins were suggested to be very critical for virus infection and entry into hosts (Abraham et al.; Tsetsarkin et al., 2009; Tsetsarkin et al., 2011b; Tsetsarkin et al., 2014). Some varieties at key sites of CHIKV E1 and E2 proteins were suggested to be involved in the virus adaption in the local mosquito species and thus influenced the spatial arrangement of different strains (Tsetsarkin et al., 2014). For example, one dominant substitution at position 226 in E1 protein (V226) was found to be related to the increasing virus fitness in A.albopictus (Tsetsarkin et al., 2007; Tsetsarkin et al., 2011b), which probably explained that the large pandemic of ECSA.IOL strains in Southeast Asia and Indian Ocean island/Indian subcontinent. No Asia.old strains were associated with epidemics after 2000, suggesting the introduction and overwhelming establishment of infection of ECSA.IOL strains in Southeast Asia for its better adaptive competence than the Asia.old strains. Subsequently, a number of ECSA.IOL strains with the substitution in E1 acquired a further mutation in E2 protein (K/Q252). The strains with the double mutations invaded more territories including South Asia and Indian Ocean islands/Indian subcontinent since 2008 than the strains with single mutation in E1 which were restricted in Indian Ocean islands/Indian subcontinent during 2005–2008 (Figure 3A). The better fitness in A.albopictus mosquito of the double mutated-ECSA.IOL strains was confirmed in laboratory by Dr. Tsetsarkin and his colleagues (Tsetsarkin et al., 2014), further supporting the roles of lineage-specific mutations in affecting virus adaption and their host spectrum and consequently the spatial and temporal distributions of different lineages. All these remind us that it is significant to monitor the genetic evolution for emergence of further adapted strains because it is very likely that amino acid variations in viral proteins would further appear to obtain more adaption to A.albopictus. The A.albopictus mosquitoes were widely distributed in the world. It has been described that they have expanded their habitats to and became abundant in the Americas and Europe thirty years before the first outbreak of CHIKV infection there (Charrel et al., 2007). Although similar to the IOL strains, the Asia.reemerge and Asia.old strains were mainly transmitted by A.albopictus that was associated with urban cycle, but were experimentally confirmed to be less adaptive to the A.albopictus mosquitoes (Tsetsarkin et al., 2007). Several specific substitutions were suggested to have epistatic effects on the virus adaption to A.albopictus endowed by the A226V mutation in E1 protein, including I211 in E2 of ECSA.Africa strains (Tsetsarkin et al., 2009) and T98 in E1 of the Asia lineage (Tsetsarkin et al.) (Figure 3A-3B), whereas the IOL strains used T211 in E2 and A98 in E1 so that the adaption ability conferred by V226 in E1 could not be blocked (Figure 3A). Moreover, the Asia.reemerge strains used A226 in E1 protein, suggesting they were less adaptive to A.albopictus (Figure 3B). Therefore, the extensive analysis of lineage-specific varieties in viral structural proteins of emerging CHIKV strains especially the proteins related to virus entry process would promote the better understanding of the functioning of viral proteins and host specificity as well as the relations of strain genotypes and distributions.
In this study, we first reported the sequences of Pakistan strains isolated from confirmed CHIKV infected human cases in the emerging epidemics in 2016, which affected over 3000 persons in Pakistan (Aamir et al., 2017). Our analyses showed that the Pakistani strains shared high similarity and all belonged to the ECSA.IOL lineage (Figure 1). Consistently, A. albopictus which was considered to be responsible for the transmission of ECSA.IOL lineage, was widely distributed and common in Pakistan (Fatima et al., 2016). However, the absence of important amino acid variants of ECSA.IOL lineage in eight strains from Pakistan might draw researchers’ attention for further surveillance. They were mostly close to the strain derived from India and migration analysis supported that the strains from Pakistan were from India (Figure 1, Figure 4). India is the neighboring country of Pakistan, where CHIKV epidemics occurred during 2006–2009, which affected a huge number of persons. However, the epidemics in India did not draw much attention from the Pakistani administration. Poor preventive and screening measures at Pakistan-India border, airports or railway stations have been performed without significant improvement after the large epidemic in India (Mallhi et al., 2017), which greatly increased the possibility of CHIKV transmission by infected travelers and mosquitoes from India to Pakistan. Further, Pakistan locates in the tropical and subtropical regions. The favorable temperate clime, ubiquitous filth and garbage and deplorable sanitary in Pakistan provide a perfect breeding place for the Aedes spp. It was reported that the Aedes spp. has been dwelling in Pakistan with a high density for a long time (Haroon and Wazir, 2016), which no doubt provided a breeding ground for the CHIKV spreading and circulating in Pakistan. There were a very few evidences suggesting that CHIKV has been existed in Pakistan years ago. In 1983, CHIKV specific antibodies were detected in sera samples from rodents and humans (Darwish et al., 1983), suggesting the possible CHIKV infection in animals and humans. In 2011, CHIKV infection were confirmed in two children during a Dengue outbreak in Pakistan (Afzal et al, 2015). We failed to know the genotypes of CHIKV strains that caused the infection in the two children as viruses were not isolated and sequenced. However, it is very possible that CHIKV has been circulating in Pakistan for years but were overlooked for insufficiency of diagnosis or misdiagnosis as Dengue virus. So the local residents in Pakistan may still expose to potential risks from CHIKV infection. The massive CHIKV pandemic in Pakistan taught a lesson to the persons working on the prevention and control of epidemic diseases in Pakistan. The analysis of the lineage-specific variations showed that the eight Pakistani did not develop the adaptive substitutions in the E1 and E2 proteins like most other IOL strains, probably because the limited numbers of Pakistani sequences were not sufficient for identifying such adaptive varieties. However, the high evolutionary rate and variations in sites of viral proteins of the IOL lineage should have drawn much attention to further characterization of the emerging CHIKV strains including the strains from Pakistan.
-
This work was supported by the Science and Technology Basic Work Program (2013FY113500) from the Ministry of Science and Technology of China, the International Cooperation on key Technologies of Biosafety along the China-Pakistan Economic Corridor (153B42KYSB20170004), the Strategic Bio-resource Service Network Plan and Building the Biogenetic Resource Preserving Capacity Program from the Chinese Academy of Sciences (ZSSB-002), and was funded by the National Basic Scientific Data Sharing-Service Platform (XXH12504-3-15).
-
The authors declare that they have no conflict of interest. The whole study was approved by the ethics committee of the Wuhan Institute of Virology (WIV), Chinese Academy of Sciences (CAS), China. Written consents were obtained from all the patients involved in the study.
-
SJ, NJ contributed the human serum sample collection; ZYS and SQL were responsible for obtaining the seven whole genomic sequence of CHIKV; ZJF and JW downloaded the sequences and constructed the datasets; JMS and SS performed the bioinformatic analysis and wrote the primary manuscript; FD, BZ and HPW checked and finalized the manuscript.
Supplementary Tables is available on the websites of Virologica Sinica: www.virosin.org; link.springer.com/journal/12250.
-
Primer pairs Sequence Length of PCR products CHIKV-F-1
CHIKV-R-1347ATGGCTGCGTGAGACACACGTAG
TTGAATGCCCATAGACARCAGC1374 bp CHIKV-F-1308
CHIKV-R-2866GAGAAAGAACACTGACCTGCTG
TTGTCTAACTGCGTAAACTCCT1559 bp CHIKV-F-2705
CHIKV-R-4151GTGGACACTACAGGCTCAACAA
GAGGGTTAGCGGCGTTGACT1447 bp CHIKV-F-3751
CHIKV-R-5302TCGCATACACCATTAYCAACAG
GTTTCTCCCTCGCCTTCTTC1552 bp CHIKV-F-5281
CHIKV-R-6813CAGAAGAAGGCGAGGGAGAAAC
AGTGAATCATCTTGGCTCTTATC1533 bp CHIKV-F-6786
CHIKV-R-8191CCTTTGATAAGAGCCAAGATGA
TGCCAGCACCTGTAGGGATG1406 bp CHIKV-F-7920
CHIKV-R-9270GCACGAAGGTAAGGTAACAGGT
CGGGACCAGAGGGGAGTTAT1350 bp CHIKV-F-9032
CHIKV-R-10430CCGAAGAGATAGAGGTACACAT
GCAGTTACAGTGATGTTATTTCC1399 bp CHIKV-F-10286
CHIKV-R-11812CTGAAAACACGCAGTTGAGC
GAAATATTAAAAACAAAATAACATCTCC1527 bp Table S1. Primers used for CHIKV genome assembly
Geographic division Country No. sequence Total WA (West Africa) Nigeria 2 14 Senegal 9 Cote dIvoire 2 Cameroon 1 SEA (South east Asia) Indonesia 4 92 Thailand 28 Philippine 7 Malaysia 11 China 17 Myanmar 5 Bangladesh 1 Cambodia 7 Singapore 12 Oceania New Caledonia 1 1 CA (Central America) Nicaragua 50 50 NA (North America) USA 45 46 Mexico 1 CI (Caribbean Island) Dominican Republic 9 10 Country 1 Saint Martin SA (South America) Brazil 6 7 French Guiana 1 ESCA (East/South/Central Africa) Congo 2 16 South Africa 3 Central African 4 Tanzania 1 Gabon 1 Uganda 1 Angola 1 Yemen 1 Kenya 2 IOL (India and Indian Ocean island) India 30 58 Sri Lanka 20 Reunion 2 Mauritius 2 Madagascar 1 Mayotte 1 Comoros 2 Pakistan_2016 Pakistan 8 8 Europe Italy 5 6 France 1 Table S2. Geographic classification of CHIKV
Extensive evolution analysis of the global chikungunya virus strains revealed the origination of CHIKV epidemics in Pakistan in 2016
- Junming Shi 1 ,
- Zhengyuan Su 1 ,
- Zhaojun Fan 1 ,
- Jun Wang 1 ,
- Siqing Liu 1 ,
- Bo Zhang 1 ,
- Hongping Wei 1 ,
- Shoukat Jehan 2 ,
- Nadia Jamil 2 ,
-
Shu Shen
1,,
 ,
, -
Fei Deng
1,,
- Received Date: 04 September 2017
- Accepted Date: 28 November 2017
- Published Date: 11 December 2017
Abstract: Chikungunya virus (CHIKV) is a mosquito-borne virus that causes epidemics widely in the world especially in the tropical and subtropical regions. Phylogenetic analysis has found that the CHIKV lineages were associated with the spatial and temporal distributions, which were related to the virus adaption to the major mosquito species and their distributions. In this study, we reported the complete genome sequences of eight CHIKV isolates from the outbreak in Pakistan last year. Then we reviewed the evolutionary history using extensive phylogenetic analysis, analyzed lineagespecific substitutions in viral proteins, and characterized the spreading pathway of CHIKV strains including the Pakistani strains. The results showed that the Pakistani stains belonged to the ECSA.IOL sub-lineage and derived from India. The genetic properties of the Pakistani strains including the adaptive substitution to vectors were further characterized, and the potential risks from the occurrence of CHIKV infection in Pakistan were discussed. These results provided better understanding of CHIKV evolution and transmission in the world and revealed the possible origination of the CHIKV outbreak and epidemic in Pakistan, which would promote the disease prevention and control in the identified countries and territories with the history of CHIKV infections as well as new regions with potential risk of CHIKV outbreaks.


 DownLoad:
DownLoad: