












HTML
-
The influenza A virus (IAV) undergoes high mutation rates and has strong seasonal transmission characteristics resulting in high morbidity and mortality in both human and animal hosts (Liu et al. 2013; Zhu et al. 2013). Deeper insights into the pathogenic mechanism of IAV are urgently required for promoting the development of vaccines and antiviral drugs. IAV belongs to the family Orthomyxoviridae and its genome consists of eight singlestranded negative sense RNA fragments. The viral RNA (vRNA), nucleoprotein (NP), and three viral polymerase proteins (PB1, PB2, and PA) form the viral ribonucleoprotein (vRNP) complex that is responsible for viral replication, transcription, and packaging (Klumpp et al. 1997; Arranz et al. 2012). The matrix protein (M1), an abundant structural component of the influenza A viral particle, acts as an essential anchor for regulating nuclearcytoplasmic shuttling of vRNP (Martin and Helenius 1991).
Ubiquitination is an enzymatic posttranslational modification that targets proteins for proteasome-mediated degradation and is tightly linked to extracellular signaling (Calistri et al. 2014). The process is orchestrated by a series of enzymes starting with the ubiquitin-activating enzyme E1, followed by the ubiquitin-conjugating enzyme E2, and finally, by E3 ubiquitin ligase that covalently links ubiquitin to the lysine (K) residue of target proteins (Chau et al. 1989; Rudnicka and Yamauchi 2016). Ubiquitination plays a vital role in IAV infection and it has been reported that the ubiquitination and de-ubiquitination of NP plays a critical role in the regulation of RNA replication of IAV (Liao et al. 2010). Furthermore, IAV RNA synthesis depends on the ubiquitin–proteasome system (Widjaja et al. 2010). There is however, very little data reported on M1 ubiquitination and its function during IAV replication.
SUMOylation is an important post-translational modification that functions similar to ubiquitination. SUMOylation of M1 at K242 ensures IAV assembly and release by forming M1-vRNP complex (Wu et al. 2011). Additionally, NS2 interaction with AIMP2 facilitates the switch from ubiquitination to SUMOylation of IAV M1 at K242 (Gao et al. 2015). E3 ubiquitin ligase Itch/AIP4 has been found to regulate M1 ubiquitination and plays important roles in IAV release from the endosome during virus entry (Su et al. 2013).
Cyclophilin A (CypA), a peptidyl-prolyl cis/trans isomerase, is an important host factor that participates in viral infection, protein folding, cell signaling, inflammation, and tumorigenesis (Handschumacher et al. 1984; Luban 2007; Sun et al. 2014). Our previous studies showed that CypA could inhibit IAV replication by promoting the RIG-I-mediated antiviral immune responses (Liu et al. 2017). Moreover, CypA interacted with IAV M1 protein and inhibited virus replication by regulating ubiquitin-proteasome degradation of the M1 protein (Liu et al. 2009; Xu et al. 2010; Liu et al. 2012). However, the precise mechanism by which CypA affects M1 ubiquitination still remains less understood. In this study, we found that E3 ubiquitin ligase AIP4 mediated M1 ubiquitination at K102 and K104 and affected M1 stability via ubiquitin-proteasome degradation pathway, while CypA inhibited M1 ubiquitination by impairing the interaction of M1 with AIP4. Furthermore, the immunofluorescence co-localization results showed that both the mutations of M1 (K102R/K104R) and CypA had strong influence on the cellular localization of M1.
-
The human embryonic kidney HEK 293 (293T), shRNAbased knockdown of CypA in human embryonic kidney 293T (CypA-/293T) described in (Liu et al. 2012), and MadinDarby canine kidney epithelial (MDCK) cell lines were grown in Dulbecco's modified eagle medium (DMEM, GIBCO, USA) supplemented with 10% fetal bovine serum (FBS, GIBCO, USA) at 37 ℃, 5% CO2. The influenza virus used in this study was the A/WSN/1933(H1N1) strain and was rescued from cDNAs and propagated in the allantoic cavities of 10-day-old specific pathogen-free embryonated chicken eggs (Beijing Merial Vital Laboratory Animal Technology Co., Ltd). Mouse anti-M1 monoclonal antibody was prepared as previously described (Koestler et al. 1984; Cao et al. 2012). Mouse anti-Flag monoclonal antibody, Mouse anti-c-Myc monoclonal antibodies, and HA-tag rabbit polyclonal antibody were purchased from Sigma; Anti-β-actin monoclonal antibody was purchased from Santa Cruz Biotechnology; Goat anti-rabbit IgG conjugated to fluorescein isothiocyanate (FITC) and goat anti-mouse IgG conjugated to tetramethyl rhodamine isocyanate (TRITC) were purchased from Zhongshan Golden Bridge Biotechnology, Beijing, China.
-
The full-length M1 gene obtained from the A/WSN/1933(H1N1) was cloned into the pCMV-Myc vector described in (Wang et al. 2013). The HA-Ub, HA-K48-Ub, HA-K63-Ub, AIP4, TRIM25, Smurf1 and CypA expression plasmids has been described previously (Liu et al. 2017). Opti-MEM was purchased from GIBCO, USA. Lipofectamine TM 2000 transfection reagent was purchased from Invitrogen. DMSO, MG132, CHX were purchased from Sigma.
-
The 293T cells were grown in 60-mm dishes in DMEM containing 10% FBS. Transfection was performed using Lipofectamine TM 2000 according to the manufacturer's instructions. After 6 h of transfection, the medium containing plasmids and transfection reagent was replaced with fresh DMEM supplemented with 10% FBS, and cells were incubated at 36 h at 37 ℃, 5% CO2.
-
The transfected cells were washed with cold phosphatebuffered saline (PBS) and the protein was extracted using cell lysis buffer (1% Triton X-100, 20 mmol/L HEPES (pH 7.4), 150 mmol/L NaCl, 1 mmol/L EDTA, 10% glycerol, complete protease inhibitor cocktail). The extracted proteins were separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. After blocking, the membranes were incubated with specific primary antibodies for 1 h at room temperature and washed three times. Next, the membranes were incubated with goat anti-mouse or anti-rabbit IgG conjugated to HRP for 1 h at room temperature and washed three times. Finally, the membranes were detected using chemiluminescence imaging system (ClinX Sciences instrument).
-
Cells were lysed and centrifuged at 11, 000 × g for 15 min at 4 ℃. The supernatants were collected and incubated with indicated antibody for 2 h, followed by the incubation of the immune complexes with Protein G agarose beads (Pierce) for at least 6 h or anti-FLAG M2 affinity gel (Sigma-Aldrich) for 4 h and subsequent 5 × 10 min washes with immunoprecipitation buffer (containing 1% Triton X-100, 20 mmol/L HEPES (pH 7.4), 300 mmol/L NaCl, 1 mmol/L EDTA and 10% glycerol). Next, the gelprotein complexes were analyzed by Western blot.
-
Cells were treated with 100 μg/mL CHX (eukaryotic protein synthesis inhibitor) for 24 h after transfection followed by lysis and subsequent analysis by Western blotting. Proteasomal blocker, MG132 (10 μmol/L) and lysosomal inhibitor NH4Cl (10 μmol/L) were used at the same time as CHX, and cells were harvested 9 h after treatment.
-
HEK 293T cells grown in 24-well cell culture plates with glass cover slides. The cells were washed with PBS three times, then fixed in 4% paraformaldehyde for 1 h at room temperature and permeabilized with PBST (0.5% Triton X-100 in PBS) at room temperature for 10 min, followed by blocking with 0.4% BSA in PBST at 37 ℃ for 1 h. Cells were then incubated with indicated antibodies diluted in PBST (containing 0.2% BSA) at 37 ℃ for 1 h. After washing with PBST for 1 h, the cells were incubated with FITC-labeled goat anti-rabbit antibody or TRITC-labeled anti-mouse antibody diluted in PBST (containing 0.2% BSA) at 37 ℃ for 1 h, then washed with PBST for 1 h. Finally, the cells were incubated with DAPI nuclear counter stain for 10 min. The intracellular location of M1 was recorded with a confocal laser scanning fluorescence microscope (Olympus LSCMFV500). All data are expressed as the means of three independent experiments ± standard deviation (SD).
-
Recombinant virus was generated with a reverse genetics system using methods described previously (Wang et al. 2013).
-
Statistically significant differences between the experimental and control groups were determined using Student's t test. P < 0.05 was considered statistically significant.
Cells, Virus, and Antibodies
Plasmids and Reagents
Transfection
Western Blot Analysis
Co-immunoprecipitation (Co-IP)
Cycloheximide (CHX), MG132, and NH4Cl Treatment
Indirect Immunofluorescence Assays (IFAs)
Generation of Recombinant Influenza A Viruses
Statistical Analysis
-
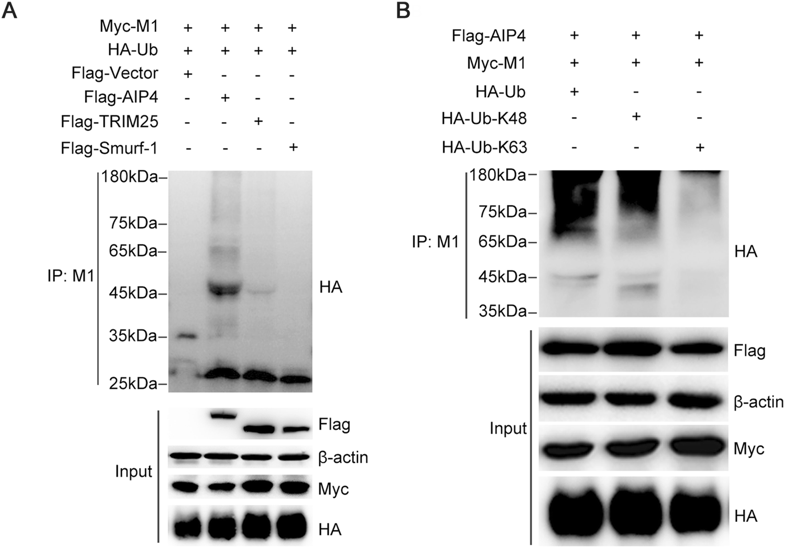
It has been reported that AIP4 is involved in M1 ubiquitination (Su et al. 2013). To investigate whether AIP4 is the specific E3 ubiquitin ligase of M1, the 293T cells were transfected with Myc-M1 and HA-Ub, along with Flagtagged AIP4 or other E3 ubiquitin ligases, such as TRIM25 and Smurf1. We observed that only AIP4 promoted M1 ubiquitination (Fig. 1A). We next examined the type of ubiquitin chain modification on M1 by AIP4. Flag-AIP4 and Myc-M1 were co-transfected with HA-Ub, HA-Ub-K48, or HA-Ub-K63 in 293T cells. As shown in Fig. 1B, AIP4 mediated K48-linked M1 ubiquitination.
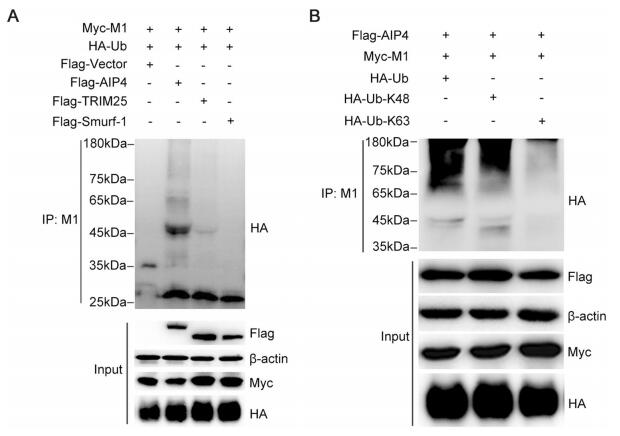
Figure 1. AIP4 mediates K48-linked ubiquitination of M1. (A) Immunoblot analysis of lysates of 293T cells transfected with Flag-vector, Flag-AIP4, Flag-TRIM25, or Flag-Smurf-1, along with Myc-M1 and HA-Ub for 36 h, followed by immunoprecipitation (IP) with anti-M1 antibody. (B) Immunoblot analysis of lysates of 293T cells transfected with HA-Ub, HA-Ub-K48 or HA-Ub-K63, along with FlagAIP4 and Myc-M1 for 36 h, followed by IP with anti-M1 antibody.
-
It is well established that the K48-linked polyubiquitin chains lead to the proteasome degradation of target protein (Finley 2009). Therefore, we examined whether AIP4 affects the stability of M1. 293T cells, co-transfected with Myc-M1 and Flag-AIP4, were treated with CHX, along with DMSO, MG132 or NH4Cl. The results showed that the proteasome inhibitor MG132 blocked the degradation of M1 promoted by AIP4, whereas DMSO and the lysosome inhibitor NH4Cl did not (Fig. 2A), indicating that AIP4 promoted the ubiquitin–proteasome degradation of M1. Additionally, we further investigated the stability of M1 regulated by AIP4 upon influenza A virus infection. Flag-AIP4 or Flag-vector was transfected in 293T cells, then infected with the influenza virus A/WSN/1933(H1N1). As expected, AIP4 promoted the degradation of M1 (Fig. 2B). Collectively, these results revealed that AIP4 promoted ubiquitin–proteasome degradation of M1.
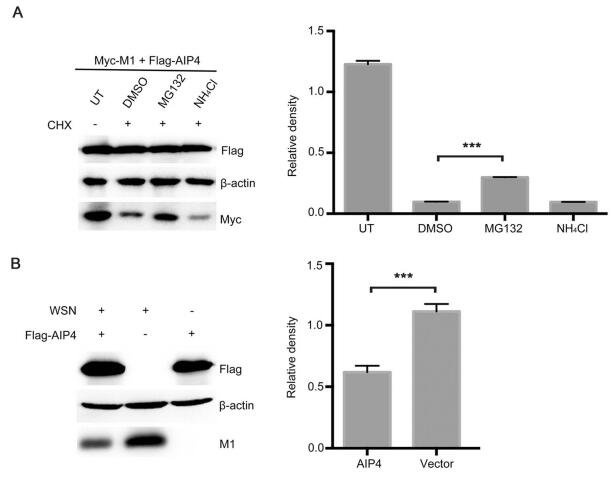
Figure 2. AIP4 affects the stability of M1. (A) Immunoblot analysis of lysates of 293T cells transfected with Myc-M1 and Flag-AIP4 for 24 h, then treated with 100 μg/mL CHX, along with 10 μmol/L MG132, 10 μmol/L NH4Cl or DMSO for 9 h (left). The relative density of M1 was normalized to β-actin (right). (B) Immunoblot analysis of lysates of 293T cells transfected with Flag-AIP4 or Flag-vector for 24 h, then infected with the influenza virus A/WSN/1933(H1N1) (MOI = 1) for 9 h (left). The relative density of M1 was normalized to β-actin (right). Data are shown as mean ± SD (n = 3). ***P < 0.001 (unpaired, two-tailed student's t test).
-
The E3 ubiquitin ligase is known to be covalently linked to the K residue of target proteins. Thus, the K residues of IAV M1 were analyzed based on the influenza sequence database (Fig. 3A). To determine the ubiquitination sites of AIP4, ten lysine residues (K21, K35, K47, K57, K95, K98, K102, K104, K113, and K187) were selected for further studies. We first constructed the M1 mutants (K to R), among which the adjacent K95/K98 and K102/K104 were constructed to have dual mutation. The 293T cells were transfected with the wild-type (WT) and mutant M1, together with Flag-AIP4 and HA-Ub. The Co-IP assay showed that only K102R/K104R blocked AIP4-mediated M1 ubiquitination (Fig. 3B), indicating that K102 and K104 are crucial for M1 ubiquitination induced by AIP4. Furthermore, we also found that the K35R mutant significantly increased the ubiquitination of M1. The precise mechanism needs to be further explored. To assess the effect of K102 and K104 on AIP4-mediated M1 stability, 293T cells were transfected with Flag-AIP4, along with WT-M1 or mutant M1 (K102R/K104R). As shown in Fig. 3C, the degradation of M1 was largely blocked by K102R/K104R, suggesting that K102 and K104 is crucial to the ubiquitin–proteasome degradation of M1 mediated by AIP4.

Figure 3. AIP4 regulates M1 ubiquitination at K102 and K104. (A) Schematic diagram representing the functional domains of M1. K21, K35, K47, K57, K95, K98, K102, K104, K113, and K187 of M1 were displayed. NEP, nuclear export protein. NLS, nuclear localization signal. (B) Immunoblot analysis of lysates of 293T cells transfected with the indicated plasmids for 36 h, followed by IP with anti-M1 antibody. (C) Immunoblot analysis of lysates in lysates of 293T cells transfected with the indicated plasmids for 36 h.
The full-length M1 sequences of 1, 429 H1N1, 195 H2N2, 1, 097 H3N2, 403 H5N1, 57 H7N7, 81 H7N9, and 437 H9N2 IAV isolates from the Influenza Virus Database of NCBI were analyzed by multiple sequence alignment. Sequence homology alignments demonstrated that the K102 and K104 residue are highly conserved among all the analyzed IAVs (Table 1).

Table 1. Conservation of K102 and K104 on M1 among different subtypes of influenza A viruses.
-
Our previous study has demonstrated that CypA is involved in the ubiquitin–proteasome degradation of IAV M1 (Liu et al. 2012), but the precise mechanism is yet to be elucidated. Since AIP4 mediates the ubiquitination of M1, we speculated that CypA might regulate AIP4-mediates M1 ubiquitination. The Co-IP assay results showed that overexpression of CypA in 293T cells inhibited M1 ubiquitination (Fig. 4A). It has been reported that both AIP4 and CypA can interact with M1 (Liu et al. 2009; Su et al. 2013). Therefore, we speculated that CypA might affect the interaction between AIP4 and M1. Co-IP assay was performed using anti-Flag beads to detect the interaction between Flag-AIP4 and Myc-M1 WT or Myc-M1 K102R/K104R in CypA-knockdown 293T cells (293T/CypA-) or CypA-overexpressing 293T/CypA-cells. We observed that CypA greatly decreased the interaction between M1 WT and AIP4, whereas the interaction between M1 K102R/K104R and AIP4 could hardly be affected by overexpressing CypA (Fig. 4B). Furthermore, we examined whether CypA regulates the stability of M1. The 293T/CypA-cells were transfected with Myc-M1 and Flag-AIP4, along with Flag-CypA or Flag-vector, then treated with CHX. The result showed that CypA suppressed AIP4-mediated M1 degradation (Fig. 4C). These results taken together suggested that CypA inhibited AIP4-induced M1 ubiquitination at K102 and K104 by inhibiting the interaction of AIP4 with M1.
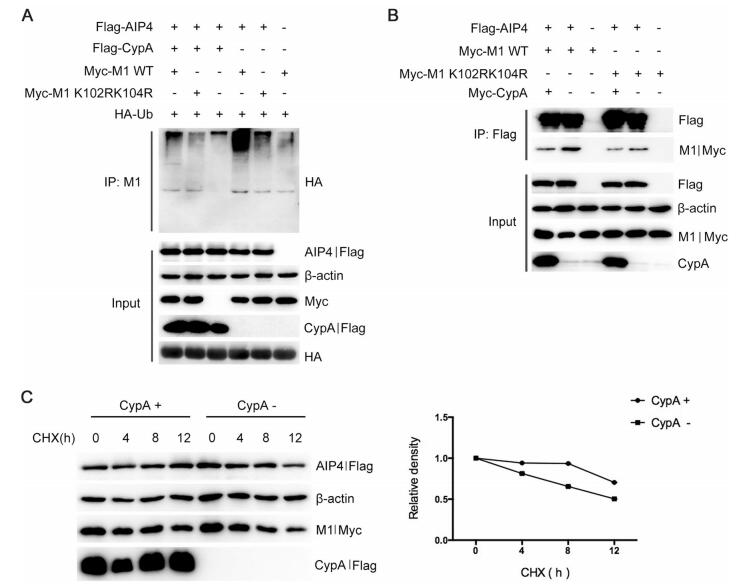
Figure 4. CypA inhibits AIP4-mediated M1 ubiquitination by impairing the interaction between AIP4 and M1. (A) Immunoblot analysis of lysates of 293T cells transfected with the indicated plasmids for 36 h, followed by IP with anti-M1 antibody. (B) Immunoblot analysis of lysates of 293T/CypA-cells transfected with the indicated plasmids for 36 h, followed by IP with anti-Flag beads. (C) Immunoblot analysis of lysates in 293T/CypA- cells transfected with Myc-M1 and Flag-AIP4, along with Flag-CypA or Flag-vector for 24 h, then treated with 100 lg/mL CHX for the indicated durations (left). The relative density of M1 was normalized to β-actin (right).
-
To investigate the effect of CypA-regulated M1 ubiquitination on the replication of IAV, we first attempted to generate a recombinant WSN virus expressing M1 K102R/K104R using the 12-plasmid reverse genetics system, but K102R/K104R did not result in any recovery of infectious virus in five independent attempts (data not shown), indicating that the ubiquitination of K102 and K104 are critical for IAV replication.
K102 and K104 are located within the NLS and NEP binding domains of M1 (Shimizu et al. 2011; Cao et al. 2012). Thus, we speculated that the ubiquitination of K102 and K104 might play a critical role in the cellular localization of M1. To examine whether CypA or K102R/K104R affect the cellular localization of M1, 293T/CypA-cells were transfected with Myc-M1 WT and Myc-M1 K102R/K104R respectively, and co-transfected with or without Flag-CypA. The results of immunofluorescence assay (IFA) showed that M1 WT displayed both nuclear and cytoplasmic localization at 12 hours post-transfection (h.p.t.), and the majority of cytoplasmic distribution at 24 and 36 h.p.t.. While M1 K102R/K104R had more nuclear distribution at all three time points than M1 WT in both 293T/CypA-cells and CypA-overexpressing 293T/CypA-cells. Additionally, M1 WT in CypA-overexpressing 293T/CypA-cells displayed greater nuclear distribution at 24 h.p.t., compared to that in 293T/CypA-cells (Fig. 5). These data revealed that both K102R/K104R and CypA resulted in nuclear retention of M1, suggesting that CypA played an important role in regulating the cellular localization of M1 via inhibiting the ubiquitination of M1 at K102 and K104.
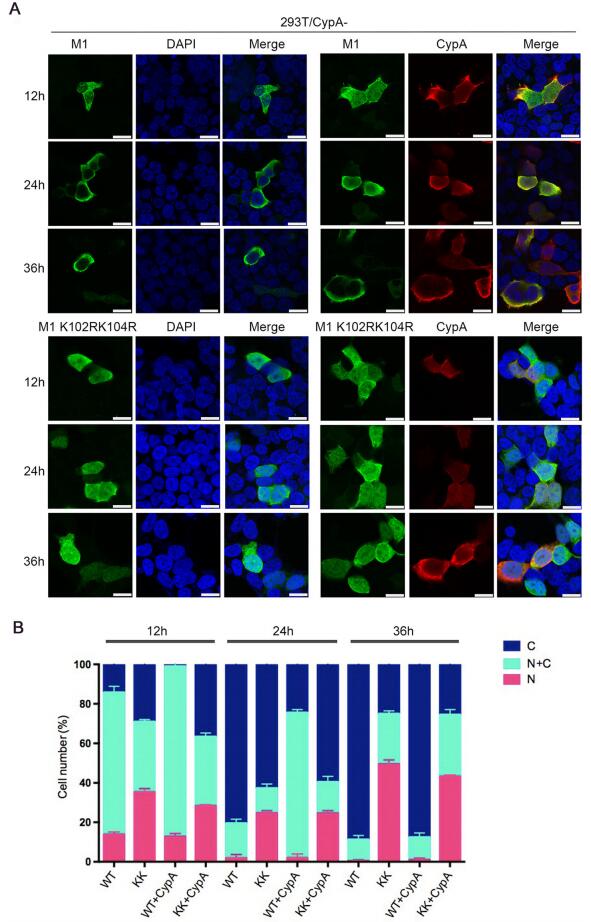
Figure 5. CypA regulates the cellular distribution of M1. (A) Cellular location of M1 determined by immunofluorescence assays. 293T/ CypA-cells were transfected with Myc-M1 WT (WT) and Myc-M1 K102R/K104R (KK) respectively, and co-transfected with or without Flag-CypA for the indicated times. The localization of Myc-M1 (green) and Flag-CypA (red) was determined using immunofluorescence assays with anti-Myc monoclonal antibody and anti-Flag monoclonal antibody. The nuclei were stained with DAPI (blue). Scale bars, 10 lm. (B) The cellular distribution of M1 in cells. At least 100 cells from each group were scored as predominantly nuclear (N), nuclear and cytoplasmic (N + C), or predominantly cytoplasmic (C), and the percentage of cells is shown. Error bars represent the standard error of the mean from at least three independent experiments.
AIP4 Mediates K48-Linked M1 Ubiquitination of IAV
AIP4 Affects the Stability of M1
K102 and K104 are Critical for AIP4-Mediated M1 Ubiquitination
CypA Inhibits AIP4-mediated M1 Ubiquitination Through Disrupting AIP4 Interaction with M1
CypA-Regulated M1 Ubiquitination Affects the Cellular Localization of M1
-
In our previous study, we had reported that CypA inhibited the replication of IAV (Liu et al. 2009). We had also attempted to elucidate the antiviral mechanism of CypA and reported that CypA increased the interaction between E3 ubiquitin ligase TRIM25 and RIG-I, promoting K63-linked ubiquitination of RIG-I. Furthermore, CypA and TRIM25 interacted with MAVS in a competitive manner, inhibiting TRIM25-mediated K48-linked ubiquitination of MAVS. Thus, CypA could inhibit virus replication via enhancing RIG-I-mediated antiviral immune responses (Liu et al. 2017). Moreover, we also found that CypA inhibited IAV replication through degradation of M1 (Liu et al. 2012). In the current study, we further elaborate that CypA inhibited AIP4-mediated ubiquitination of M1 at K102 and K104 by impairing the interaction of AIP4 with M1. It is possible that the ubiquitination of M1 as one of the modifications to modify target proteins can improve the IAV replication by inhibiting the functions of M1, which played a major role in viral replication. As the degradation of M1 is only one of the outcomes M1 ubiquitination, its role in inhibiting viral replication may only be a minor one. The above studies reveal a ubiquitination-based mechanism by which CypA regulates the replication of IAV.
Ubiquitination, a dynamic and reversible post-translational modification that plays a vital role in cellular homeostasis. During their evolution, many viruses attempt to escape immune responses of the infected host cells. They can establish a successful infection by hijacking the ubiquitination pathway of the host. For example, the influenza virus selectively exploits the ubiquitin–vacuolar protein sorting system to entry into the host cells successfully (Khor et al. 2003). Compared with the phosphorylation of influenza virus proteins, there are few studies focusing on their ubiquitination. K184 monoubiquitination of nucleoprotein (NP) increases IAV RNA replication and cellular deubiquitinating enzyme USP11 decreases polymerase activity and viral replication. (Liao et al. 2010). Ectopically overexpressing ubiquitin up-regulates polymerase function in a dose-dependent fashion causing increased accumulation of vRNA, cRNA and mRNA and enhanced viral gene expression during infection (Kirui et al. 2016). E3 ligase TRIM22 was reported to inhibit influenza A virus infection by targeting NP for degradation (Di Pietro et al. 2013). AIMP2 interacts with nonstructural protein 2 (NS2, also named NEP) of IAV and suppresses M1 ubiquitination at K242 and protein degradation by an unknown mechanism, and then promotes IAV replication (Gao et al. 2015). Collectively, ubiquitination can positively or negatively regulate viral replication via different mechanisms. Here, we showed that CypA impaired AIP4-mediated ubiquitination and degradation of M1 and reduced the replication of IAV, suggesting that the ubiquitination and degradation of M1 played a positive role during viral replication. Furthermore, as shown in Fig. 2B and Fig. 3C, the K102R/K104R mutations resulted in dramatic but not complete inhibition of M1 degradation, indicating that there may be other ways in which M1 undergoes degradation. We speculated that other E3 ubiquitin ligases and ubiquitination sites on M1 might participate in the ubiquitination of M1 and play different roles. Further studies on the ubiquitination mechanism of IAV proteins may yield exciting results.
At the early stage of IAV infection, newly synthesized M1 translocates to the nucleus to mediate vRNP nuclear export (Ye et al. 1987; Sha and Luo 1997; Bui et al. 2000; Baudin et al. 2001). While at the late stage, M1 is exported from the nucleus to block the reentry of vRNPs into the nucleus, mediate viral assembly and budding, and control virus morphology (Martin and Helenius 1991; Burleigh et al. 2005; Calder et al. 2010; Wu et al. 2011). In this study, we observed that CypA and K102R/K104R altered the cellular localization of M1. It has been reported that K102 and K104 are located within the NLS and NEP binding domain of M1, and NEP binding to M1 is essential for the nuclear export of vRNP in infected cells (Shimizu et al. 2011). Therefore, it is possible that CypA could regulate the nucleocytoplasmic shuttling of vRNP by affecting the NLS of M1 and/or NEP binding to M1, which need to be further explored.
In summary, we show that CypA inhibited AIP4-mediated M1 ubiquitination at K102 and K104 by impairing the interaction of AIP4 with M1, followed by the suppression of the nuclear export of M1, and finally reduction of the replication of IAV. Essentially our study provides deeper insights of the ubiquitination regulation mechanism of IAV proteins and replication. Studies such as this pave the way for more sophisticated therapeutic approach to combat influenza infection.
-
This work was supported by grants from the National Natural Science Foundation of China (31630079, 31672531, 31572526, and 31802164), the National Key R & D Program of China (2016YFD0500206, 2015BAD11B02), the Strategic Priority Research Program of Chinese Academy of Sciences (XDB29010000), the National Science and Technology Major Project (2018ZX10101004), and the Emergency Technology Research Issue on Prevention and Control for Human Infection with A (H7N9) Avian Influenza Virus (10600100000015001206). Wenjun Liu is the principal investigator of the Innovative Research Group of National Natural Science Foundation of China (Grant No. 81621091).
-
LS and Wenjun Liu supervised the project, designed the study, and analyzed the data; MM and LS were responsible for planning and conducting the experimental work, and wrote the manuscript; WZ, Wei Liu and WF provided the technical support; LC, YL, PJ and WY participated in part of experimental work. JL and LY analyzed the data.
Acknowledgements
Author Contributions
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.


 DownLoad:
DownLoad: