














HTML
-
Dengue fever (DF) is an arthropod-borne tropical acute infectious disease caused by dengue virus (DENV) that is transmitted to humans primarily by Aedes aegypti and Aedes albopictus mosquitoes (Lustig et al. 2017). The most common clinical symptoms of the infected patients are high fever, rash, lethargy, joint pain, leucopenia, and lymphadenopathy (WHO 2009). Furthermore, according to the revised WHO dengue classification scheme, dengue patients who recover without major complications are classified as having dengue (Srikiatkhachorn et al. 2010). Dengue patients who experience dengue hemorrhagic fever, shock syndrome, an accumulation of serosal fluid sufficient to cause respiratory distress, severe organ impairment, or death are classified as having severe dengue (Srikiatkhachorn et al. 2010).
DENV belongs to the genus Flavivirus of the family Flaviviridae. Four serotypes of DENV have been identified (DENV-1 TO DENV-4) (Normile 2013; Anez et al. 2017). Of these serotypes, DENV-1 exhibits extensive diversity and has been divided into five distinct genotypes: genotype Ⅰ (Southeast Asia and East Africa); genotype Ⅱ (Thailand); genotype Ⅲ (Malaysia); genotype Ⅳ (South Pacific); and genotype Ⅴ (America/Africa) (Holmes and Twiddy 2003). Among these genotypes, genotype Ⅰ is further divided into four lineages (lineages Ⅰ to lineages Ⅳ) (Zheng et al. 2009; Chen 2011; Bai et al. 2018). The DENV genome is a single-stranded, positive-sense RNA virus approximately 10, 000–11, 000 nucleotide bases in length. The DENV genome also contains a single open reading frame encoding a polyprotein, as well as 5′ and 3′ untranslated regions (UTRs). The polyprotein consists of three structural proteins (capsid [C], membrane [PrM/M], and envelope [E]) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (Chambers et al. 1990).
DF is an epidemic disease characterized by outbreaks primarily in tropical and subtropical areas, such as the south America, southern Europe, southeast Asia, western Pacific, north Africa, Australia, the eastern Mediterranean, and the Indian Ocean islands (Brady et al. 2012). In recent years, the spread of the DENV has led to a rapid increase in the morbidity of DF. In particular, DF cases have been reported in more than 100 countries, and approximately 390 million people are infected per year, including 96 million cases of severe dengue (Bhatt et al. 2013). In China, the first DF case appeared in Xiamen City, Fujian Province in 1873, which was followed by an epidemic in the Shanghai, Zhejiang of the southeast coastal area. In 1978, imported DENV-1 and DENV-4 caused an outbreak of DF in Foshan City, Guangdong Province (Qiu et al. 1993). In 1980, DF was epidemic in Hainan Province and both DENV-1 and DENV-3 were isolated from patient serum (Li et al. 1986). In 1982, DENV-2 caused an epidemic outbreak in the northern area of Hainan Province (Qiu et al. 1991). In 2004, DENV-1-infected patients were first discovered in Ningbo City and the viral genome exhibited a high sequence identity with that of DENV-1 from primarily pandemic strains in Thailand. This indicated that infected travelers from Thailand introduced the strains into China (Xu et al. 2007). Since 1990, DF continued to exist in Guangdong and Guangxi Provinces. However, in 2014, Guangdong Province suffered the worst DF epidemic in its history. The outbreak was caused by DENV-1 ~ 4 and the number of DF cases was 10 times greater than that reported in 2013 (Yu et al. 2019). According to the laboratory diagnosis, DENV-1 was the major causative agent in the DF outbreak (Xiao et al. 2016).
Over the past two decades prior to 2019, no dengue outbreak was reported in Hainan Province. However, in early September of 2019, an increasing number of DENV-infected cases appeared, and the DF outbreak lasted for over a month in Haikou City, Hainan Province. In this study, we collected 97 plasma samples from DF patients from five hospitals and 1585 mosquito larvae samples from the puddles in different areas of Haikou, isolated the pathogens and investigated the genome characteristics of DENV. Result showed that the DENV could be detected both in the patient's plasma samples and mosquito larvae samples, and the new strains were closely related to the epidemic strain in the southern regions of China. Our results indicate that the DENV causing the 2019 DF outbreak in Hainan Province may have been imported from Chinese mainland and formed a transmission cycle between humans and mosquitoes.
-
The inclusion criteria consisted of patients who were diagnosed with DF in Hainan Province according to the National hygienic standard (WS 216-2018). A total of 97 plasma samples were collected from 97 DF suspected patients from Second Affiliated Hospital of Hainan Medical University, Haikou Municipal Hospital and Hainan Maternity and Child Care Hospital during the DF outbreak from September 11 to October 17, 2019. The infected sites of those DF patients were located throughout Haikou City, including the Wuyuanhe Community and Shuchang Village in Xiuying District, the location in which the original DF patient was reported. Whole blood samples were collected in an ethylene diamine tetraacetic acid (EDTA) blood collection tube. After the whole blood samples were centrifuged, the upper plasma samples were transported to the laboratory within 24 h using cold chain transportation and stored at -80 ℃.
-
The Aedes albopictus larvae samples were collected from the puddles in dumps located in Xiuying and Longhua District. There are two sampling points A and B in Xiuying District and one sampling point C in Longhua District (Fig. 1). A total of 1585 Aedes albopictus larvae were divided into 53 pooled samples, including 33 mixed samples (994 mosquito larvae) from Point A, 19 mixed samples (570 mosquito larvae) from Point B and 1 mixed sample from Point C. The pooled samples were homogenized and the viral nucleotides (including both the viral genome DNA and viral mRNA) were isolated using a QIAamp® Viral RNAMini Kit (Qiagen, Germany). Serotype-specific primers were used to identify the nucleic acid sequence of the E protein by PCR amplification and Sanger sequencing (Yu et al. 2019). The larval species was determined morphologically (Supplemental Fig. S1) and confirmed by a DNA sequence analysis of mitochondrial cytochrome b.
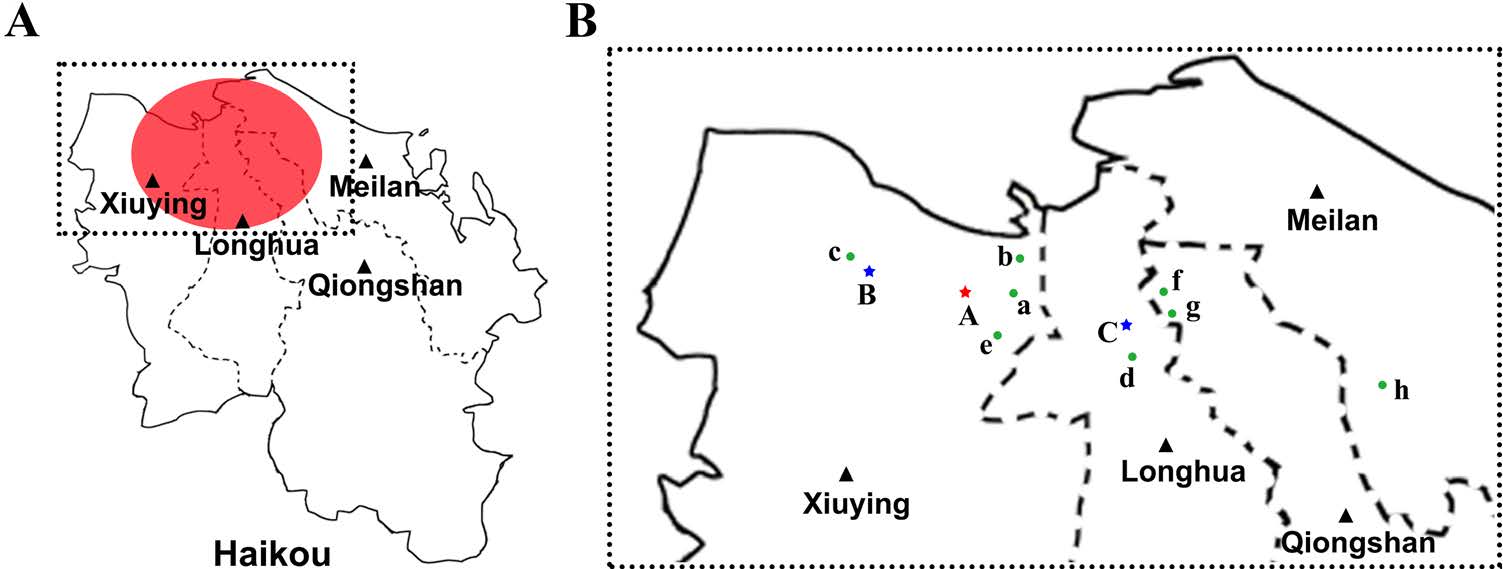
Figure 1. Outbreak regions of the DF and the sampling sites in Haikou 2019. A Outbreak regions of the DF. Red circle: Outbreak regions of the DF in Haikou 2019. B The mosquito larvae sampling sites and patient locations. Point (A–C): Mosquito larvae sampling sites. Two sampling points (Point A and B) in Xiuying District and one sampling point (Point C) in Longhua District. Green dots (a–h): The location of eight patients. Blue star: The location of the negative mosquito larvae samples. Red star: The location of the positive mosquito larvae samples.
-
Immunochromatography and the double antibody sandwich method were used to qualitatively detect the DENV NS1 antigens in the plasma samples of 97 individuals (colloidal gold probes kit, Wantai, Beijin, China). The viral RNA was extracted from 140 μL plasma samples using the QIAamp® Viral RNAMini Kit (Qiagen, Germany). A superscript Ⅲ first-strand cDNA Synthesis Kit (Invitrogen, USA) was used for the transcription of DENV RNA. Nested polymerase chain reaction (PCR) was used to amplify E gene cDNA using the GoTaq® Long PCR Master Mix (Promega, USA). Serotype-specific primers were used to confirm the serotype of randomly selected colloidal positive samples from the different infected sites (Yu et al. 2019) (Supplementary Table S1). The first round of PCR was primed with outer primers and the second round PCR was primed with inner primers. A total of 1 μL of the first round PCR product was used as the template for the second round of PCR. The thermal cycling conditions for both PCR rounds were 94 ℃ for 5 min, followed by 35 cycles of 94 ℃ for 30 s, 52 ℃ for 30 s, 72 ℃ for 2 min, and a final elongation step at 72 ℃ for 10 min. Finally, the PCR products were analyzed via 1.5% agarose gel electrophoresis ultraviolet imaging. The amplicons were randomly selected for Sanger sequencing for confirmation. The representative DENV strain was selected to determine and characterize the complete genome sequence (Supplementary Table S1). Eight targeted amplicons with overlapping sequences were assembled into a complete genome sequence.
-
The amino acid sequences of the ORFs were deduced by comparing the sequences with those of other DENV strains. The conserved protein families and domains were predicted using Pfam and InterProScan 5 (http://www.ebi.ac.uk/services/proteins). Routine sequence alignments were performed using Clustal Omega, Needle (http://www.ebi.ac.uk/Tools/) (Zhu et al. 2016).
-
MEGA6.0 was used to align the nucleotide sequences and deduce amino acid sequences with the MUSCLE package and default parameters. The phylogenetic tree was constructed via the maximum-likelihood method based on the complete nucleotide sequences of the envelope gene regions. The neighbor-joining method was used as a substitution model that was carried out using the model selection function of MEGA6.0 with 1000 bootstrap replicates. The tree was drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The rate variations among the sites were modeled with a gamma distribution (shape parameter = 1). Nucleotide pairwise alignment between the HMU-HKU and other members of DENV were performed with the NCBI Basic Local Alignment Search Tool. The translated amino acid sequences were analyzed by BioEdit7.1.3.
Patient Plasma Samples
Mosquito Larvae Samples
Epidemiological Data and Viral Genome Sequencing
Genome Annotation
Analysis of the Phylogenetic and Molecular Characteristics
-
The Aedes albopictus clone C6/36 cell line (C6/36; ATCC, Manassas, VA) were propagated and maintained in Dulbecco's Modified Eagle Medium (HyClone, Logan, UT) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) plus 100 U/mL penicillin and 100 μg/mL streptomycin at 28 ℃ with 5% CO2. The total number and proliferating cells were determined by direct cell number counting using CountStar (Inno-Alliance Biotech, Beijing, China) after staining with trypan blue. Acute phase serum from the patients was collected, diluted 1:100, and inoculated onto C6/36 cells in 6-well plate.
-
The DF epidemics occurring in Haikou, in the northern region of Hainan islands were the closest to the mainland of China. The DF outbreak began on September 5th, 2019 and lasted until the end of October. More than 200 infected patients were reported as being infected with DENV according to the Hainan Province Department of Health. There was neither severe dengue nor death among the DENV-infected individuals. Infection primarily occurred in the Xiuying District, and the remaining cases were distributed in the districts of Meilan, Qiongshan, and Longhua in Haikou City, Hainan Province (Fig. 1).
-
The patients were diagnosed as dengue virus infection in the hospital by fluorescence quantitative RT-PCR (ShuoShi/Jiangsu, China) targeted with 3′ Untranslated Regions (3′-UTR). All of 97 DF cases are mild and these patients showed a good prognosis. The common feature of these DF patients were fever, fatigue, headache, flushing, muscle aches and joint pain. The DENV NS1 antigen was tested in 97 plasma samples using colloidal gold probes based on fluorescent immunochromatography. The DENV NS1 and colloidal gold binding antibodies were prepared by gene engineering. There were 49 of the 97 plasma samples found to be strongly positive and 9 of 97 the plasma samples were weakly positive against the NS1 antigen (Supplementary Table S2). Figure 2 shows that number of patients ranged from 5 to 24 in the different age groups. Despite the different number of patients in each group, the proportion of colloidal gold-positive patients was significantly enhanced in the age groups of 41–50 and 51–60, and were very low in the age groups of < 1 and 1–10.
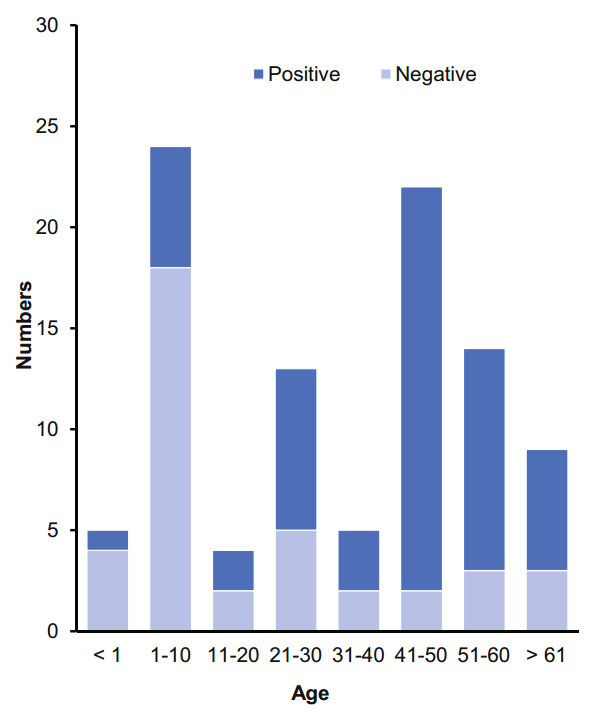
Figure 2. The plasma positive rate of the colloidal gold method in different age groups. The 97 plasma samples from 97 suspected DF patients of different ages. The number of positive samples are labeled in dark blue and the number of negative samples are labeled in light blue.
The 53 pooled samples of Aedes albopictus larvae were screened by serotype-specific primers of DENV, of which only one sample from Point A in Xiuying District was positive (Fig. 1). Furthermore, PCR amplification of E protein sequencing was verified by Sanger sequencing as DENV-1, genotype Ⅰ (DENV-1 HMU-HKU-9, GenBank accession number: MW206000).
-
Since Xiuying District of Haikou City has the most confirmed DENV infected cases, followed by Qiongshan District, then Meilan District and Longhua District, we randomly collected eight plasma samples for complete E protein sequencing (four plasma samples from Xiuying District, two from Qiongshan District, one from Meilan District and Longhua District, respectively) (Fig. 1). Those sequences from plasma samples (DENV-1 HMU-HKU-1 to DENV-1 HMU-HKU-8, GenBank accession number: MT447146–MT447153) and the sequence from mosquito larvae sample (DENV-HMU-HKU-9), were aligned and compared with 123 representative sequences of diverse geographical origin, including four serotypes of DENV, five genotypes of DENV-1, and four lineages of Genotype-1 acquired from the NCBI GenBank database. Evolutionary trees were constructed from the complete nucleotide sequences of the E protein (Fig. 3). The topologies of the tree showed that DENV1 HMU-HKU clustered with lineage Ⅳ, which represented an independent rooted branch belonging to serotype 1, genotype Ⅰ (Fig. 3). The nine Hainan isolated strains that were closely located in one cluster were close to the Ningbo strain of China (2018, MK203786 and MK764265). Furthermore, the nine isolated strains were also closely related to the Zhejiang strain (2017, MH010601), Guangdong strain (2017, MK566217), Yunnan strain (2017, MF683117).

Figure 3. DENV phylogenetic tree. A phylogenetic tree based on the complete envelope protein sequences of each DENV1 HMU-HKU. The viruses found in this study are labeled with a red circle (

The DENV-1 HMU-HKU-2 strain isolated from the patient plasma samples was selected for amplification and characterization of the whole genome sequence. Result shows that DENV-1 HMU-HKU-2 has the typical genome organization of the genus Flavivirus and has 98%-99% nucleotide identity with other serotype 1, genotype Ⅰ, lineage Ⅳ DENVs.
-
The acute phase serum from the patients who were clinically diagnosed as DF was further validated by PCR with DENV primers (Yu et al. 2019) (Supplementary Table S1). The 40 µL serum of Each PCR-positive samples was inoculated onto C6/36 cells in 6-well plate. The cells were propagated for 7 days until clear cytopathic effects (CPE) were observed. Membrane fusion and cytoplasm shrinkage appeared after 7 days and had worsened after 10 days of DENV infection (Fig. 4). There were no observable morphological changes in the negative controls. Three samples induced complete CPE, and finally two strains of virus were isolated successfully.
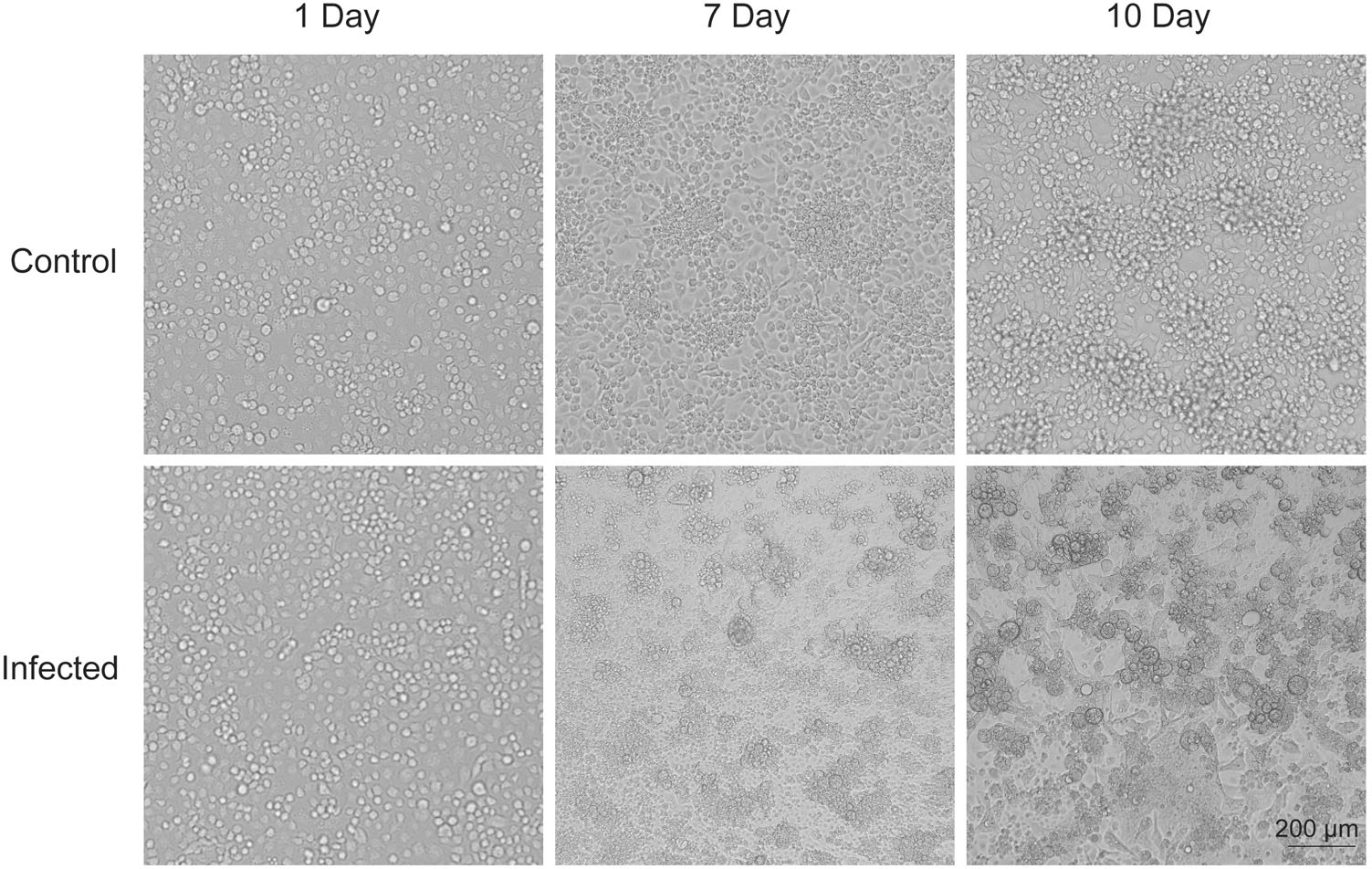
Figure 4. C6/36 cells exhibited cytopathic effects. C6/36 cells were evaluated for cytopathic effects at 1 d, 7d, and 10 d after inoculation with acute phase serum from the patients. Membrane blebbing and cytoplasm shrinkage appeared after 7 d and worsened after 10 d of DENV infection. No morphological changes was observed in the negative controls.
-
The base substitution and amino acid mutation analyses were performed by comparison with the DENV-1 prototype strain (EU848545, US/Hawaii/1944). The results showed 144 base substitutions in the structural region (C, PrM, M, and E) of HMU-HKU-2, of which 36 substitutions were non-synonymous. There were 475 base substitutions in the non-structural regions in the HMU-HKU-2, leading to 63 non-synonymous mutations. A total of 90 amino acid mutations occurred in the CDS of two strains (US/Hawaii/1944 and HMU-HKU-2).
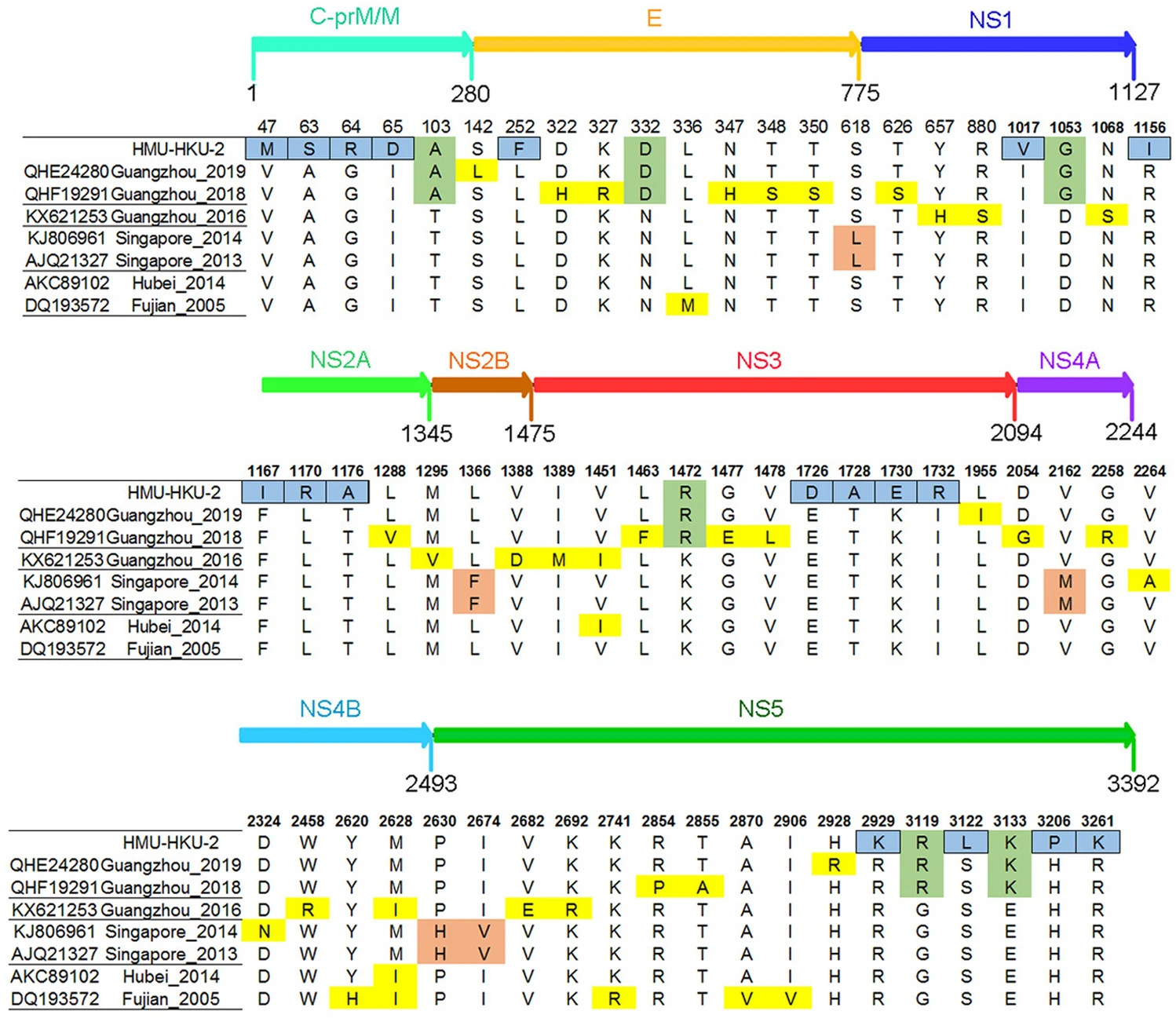
According to the phylogenetic analysis and NCBI Basic Local Alignment Search Tool (BLAST), seven complete DENV-1 amino acid strains were the most closely related to HMU-HKU-2. When compared to the seven closest strains, a total of 18 amino acid mutations were predicted in the CDS of HMU-HKU-2 (Fig. 5). Of these, five amino acid mutations (V47M, A63S, G64R, I65D, and L252F) occurred in the structural protein C-prM/M and E, five amino acid mutations (I1017v, R1153I, F1167I, L1170R, and T1176A) in the non-structural proteins, NS1 and NS2, and eight amino acid mutations (E1726D, T1728A, K1730E, I1732R, R2929K, S3122L, H3206P, and R3261K) occurred in the non-structural proteins, NS3 and NS5. The amino acids changed from polar to nonpolar residues at positions R1156I, T1176A, T1728A, S3122L, and H3206P, whereas the changes were from nonpolar to polar residues in A63S, I65D, L1170R, and I1732R.
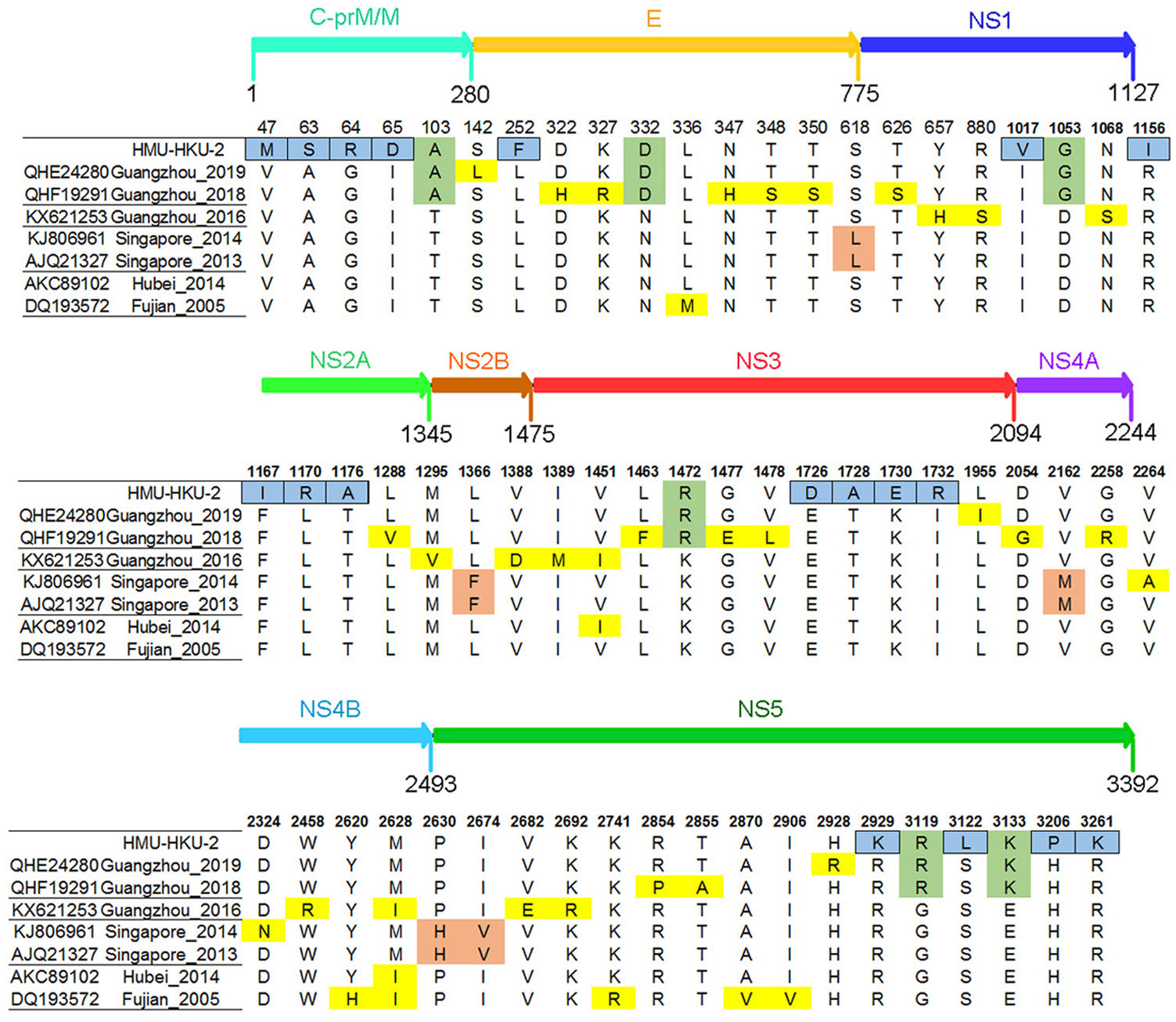
Figure 5. Amino acid mutation analysis of HMU-HKU-2. The amino acid mutations in the CDS of the HMU-HKU-2 are labeled with a blue square. The amino acid mutations that occurred in the CDS of Guangzhou 2019, Guangzhou 2018, and HMU-HKU-2 are labeled with a green square.
Geographic Analysis of the Outbreak
Epidemiological Data
Phylogenetic Analysis
DENV-Infected C6/36 Cells Exhibit Cytopathic Effects
Base Substitution and Amino Acid Mutation Analysis of HMU-HKU-2
-
Currently, DF is an imported infectious disease and there is no obvious natural epidemic foci of dengue virus that exists in China. From 1978 to 1991, DF was primarily epidemic in the Guangdong and Hainan provinces in China (Qiu et al. 1993). The morbidity of DF in Hainan Province was the highest in China from 1978 to 1992 (Li et al. 1986); however, there have been a few DF reported cases during the past two decades in Hainan Province. After 1990, there was an increased number of DF reports in Guangxi, Fujian, Yunnan, Hong Kong, and Zhejiang (Xu et al. 2007). From 2005 to 2014, the DF outbreaks were reported nationwide and the number of DF cases in Guangdong, Zhejiang, Fujian, and Yunnan provinces accounted for more than 90% of the total DF cases in Chinese mainland (Wu et al. 2010). From 2014 to 2018, the DF cases were gradually distributed from southwestern and southern regions to the southeastern coastal parts of China, and subsequently to the central and northern regions of China (Yue et al. 2019). While DENV-1 ~ 4 were alternately epidemic in Chinese mainland, DENV1 was predominant. Recently, the major serotype, DENV-1, has been epidemic in Guangdong for several consecutive years.
Among the five genotypes of DENV1, genotype Ⅰ has circulated extensively in the southern and southeastern provinces of China. The molecular evolutionary analyses revealed that Genotype Ⅰ has branched into four lineages. Jianhai Yu et al. described the circulation of these four lineages in Guangdong Province from 2006 to 2016 (Yu et al. 2019). Lineage Ⅰ was epidemic from 2006 to 2010, lineage Ⅱ from 2006 to 2012, lineage Ⅲ from 2006 to 2015, and lineage Ⅳ from 2007 to 2016. In our phylogenetic analysis, the sequences of the four DENV-1 genotype Ⅰ lineages were obtained from Genebank, including the sequences from the mainland of China and those of neighboring countries (e.g., Indonesia, Malaysia, Thailand, and Singapore). The genotypes Ⅱ-Ⅴ of DENV-1 and DENV-2 ~ 4 were used as outgroup. Eight newly isolated Hainan strains have a close relationship with the epidemic strains in southern and southeast province of China isolated in 2017 or 2018. Along with the strains circulating in neighboring countries, these strains formed a large cluster belonging to lineage Ⅳ. Our data indicated that the 2019 DF outbreak in Hainan Province was likely due to infected mosquitos or patients imported from Chinese mainland rather than from the neighboring countries.
In addition, in some special sites, the amino acid mutations have had a crucial impact on the replication and virulence of flaviviruses, including mutations of putative N-glycosylation (Asn-58 and Asn-62) sites in DENV NS4B, which affects the biological function of viral replication (Naik and Wu 2015). The PDK53 (vaccine strain) has three critical mutations for the attenuated state of PDK53 (Butrapet et al. 2000). In this study, the complete amino acid sequence of HMU-HKU-2 was found to share the highest identity with the Guangzhou_2019 and Guangzhou_2018 strains based on the BLAST and amino acid mutation analysis (Fig. 4). It is interesting that the continuous mutation of three amino acids occurred in the capsid protein C region of HMU-HKU-2 (A63S, G64R, and I65D), and the continuous four amino acid mutations by one site interval each occurred in the flavivirus DEAD domain region of HMU-HKU-2 (E1726D, T1728A, K1730E, and I1732R) (Kumar et al. 2017). DEAD-box RNA helicases play an indispensable role in a wide-range of metabolic processes (Jeske et al. 2017). However, whether those amino acid substitutions in the capsid protein C region and the flavivirus DEAD domain region are affected on replication, infection and other biological functions still unclear.
Furthermore, it is interesting that the colloidal gold-positive proportion of the patient's plasma samples was substantially higher in the 41–50 and 51–60 age groups compared to that in the < 1 and 1–10 age groups based on the 97 plasma samples. However, this is different from other reports that children were the most vulnerable age group. The result requires further study based on the large quantity of samples from a diverse number of infected sites. In addition, among the 53 pooled samples of the total 1585 Aedes albopictus larvae, only one pooled sample was positive for DENV-1. In phylogenetic analysis, DENV-HMU-HKU-9 and DENV-HMU-HKU-1 ~ 8 were closely related, and they were located in one cluster with 99.84%–100% nucleotide identify with each other. These findings implied that in this 2019 DF outbreak, DENV-1 was able to vertically transmit from the mosquito to the mosquito larvae and formed a transmission cycle between humans and mosquitoes in Hainan Province, suggesting that regular molecular epidemiological surveillance is quite necessary for control and prevention of DENV transmission.
-
This work was supported by the Hainan Provincial Natural Science Foundation of China (Grant No. 2019RC218), the National Natural Science Foundation of China (grant No. 81860367, 31460017), the Scientific Research Foundation of the Higher Education Institutions of Hainan Province, China (Grant No. Hnky2020- 33), the Hainan Medical University novel coronavirus pneumonia project (Grant No. XGZX2020005), the Hainan Provincial Natural Science Foundation of China (Grant No. 819QN360), State Key Laboratory of Virology 2018 Open Fund Project (2018IOV002), the National S & T Major Project "China Mega-Project for Infectious Disease" (Grant No. 2018ZX10711001), and the CAMS Innovation Fund for Medical Sciences (Grant No. 2017-I2M-3-017).
-
FY designed the study. YD, MX, LZ, XH, RP, GW, YH, WW, KW, QW, XC, LN, YZ, CT, GL collected the specimens and performed theexperiments. JD, FY, HS, FYanalyzed thedata. JD, F Y wrote the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This study is approved by the Ethics Committee of the Hainan Medical University. The research content and significance of our study were explained and written informed consents were obtained by all the dengue patients and patient's guardians. All experiments were performed in accordance with the experimental guidelines for molecular biology, and abide by biosafety regulations. All methods involving humans were performed in accordance with the guidelines and regulations of the Declaration of Helsinki.


 DownLoad:
DownLoad: