













HTML
-
Viral myocarditis (VM) is an inflammatory disease of the myocardium associated with the development of heart failure and sudden death in young and previously healthy individuals (Corsten et al. 2015; Li et al. 2016; Rienks et al. 2017). According to available clinical evidence, the mortality of VM in young people is as high as 21%, and sudden deaths due to VM or fatal ventricular arrhythmias in children account for about 20% of deaths (Ma xrquezGonza xlez et al. 2016; Yu et al. 2016). Many viruses can cause VM, including coxsackieviruses, adenoviruses, influenza viruses, cytomegaloviruses, and human immunodeficiency virus (Grist and Reid 1997), coxsackievirus B3 (CVB3) accounts for a major portion of VM cases. It has been reported that 1/4 of dilated cardiomyopathy and myocarditis in children and young patients is caused by CVB3 (Liao et al. 2015; Esfandiarei and McManus 2008). CVB3 is known to cause direct cardiomyocyte injury, and cardiac inflammation underlies CVB3-induced viral myocarditis (Yuan et al. 2009; Van Linthout et al. 2011). However, the mechanism of CVB3-induced myocarditis remains unclear.
MicroRNAs (miRNAs) are small non-coding RNAs, containing approximately 22 nucleotides, that can posttranscriptionally regulate gene expression by interacting with target messenger RNAs (mRNAs) (Cullen 2006; Tong et al. 2013). They act as the important regulators of gene expression and influence a wide variety of biological processes, including cardiac biology and regulation of immune responses (O'Connell et al. 2010; Small and Olson 2011). CVB3 infection causes dysregulation of non-coding RNAs, like miRNAs, lncRNAs, which is characteristic of VM (Zhang et al. 2020). Earlier research has shown that miRNAs regulate viral replication by targeting the mRNA of host cells or viral genome. miR-126 enhances CVB3 replication by targeting SPRED1, LRP6 and WRCH1 genes (Ye et al. 2013), while miR-342-5p reduces CVB3 replication by targeting the 2C-coding region (Wang et al. 2012). Also, many miRNAs have been shown to have a significant effect on the immune response and inflammation associated with myocarditis. In other researches, miR-155 inhibition was found to reduce mortality and improve cardiac function in CVB3-induced VM by regulating monocyte-macrophages and T lymphocyte activation (Corsten et al. 2012). LncRNAs belongs to non-coding RNAs with a length longer than 200 nucleotides. LncRNAs could regulate miRNAs to influence the CVB3 replication, like lncRNA HIF1A-AS1 targeting miR-138 to aggravates CVB3-induced myocardium inflammation, lncRNA AK085865 regulates of macrophage polarization by targeting miR-192 (Cao et al. 2020; Zhang et al. 2020). Circular RNAs (circRNAs) are noncoding RNAs obtained by back-splicing of precursor mRNAs from exons of multiple eukaryotic genes. Many researches showed that, miRNAs played an important role on viral myocarditis, but the mechanism by which miRNAs regulated viral replication and functions in CVB3-initiated myocarditis remained unknown.
The tripartite motif (TRIM) family is a diverse family of RING finger domain-containing proteins, including more than 70 members in humans (Nisole et al. 2005; Meroni and Diez-Roux 2010; Kawai and Akira 2011). TRIM27 is a member of the TRIM family, which is involved in various cellular processes, including cell proliferation, differentiation, apoptosis, autophagy, and innate immune responses (Hatakeyama 2017). It activates MAPK-and NF-jBdependent genes and significantly contributes to the regulation of immune responses. In recent years, TRIM27 has been shown to regulate the innate antiviral immune responses against the infection of vesicular stomatitis virus, herpes simplex virus, Sendai virus, and hepatitis C virus (Zheng et al. 2015a, 2015b, 2019). And some researches showed miRNAs could target TRIM27 for playing their role. miR-183 cluster regulates the innate antiviral response by target TRIM27 (Singaravelu et al. 2019). And miR-27a acts as the negative regulator of type I interferon (IFN) production for regulating vesicular stomatitis virus replication in macrophages (Zheng et al. 2015a). Many studies have shown that TRIM27 plays an important role in host antiviral responses, but only a few of them focused on the relationship between TRIM27 and CVB3-induced VM. In our study, CVB3 infection reduced the TRIM27 expression, and by binding site prediction and luciferase assay, the miR-324-3p could target TRIM27 for playing its function.
In the previous study, we found miR-324-3p had a significantly differentially expression in mice cardiac tissues by miRNA microarray, so we investigated the roles of miR-324-3p in CVB3 replication and CVB3-induced VM. Our study revealed that miR-324-3p played a negative role in CVB3 infection by targeting CVB3 replication, regulating the viral load, and reducing the inflammatory response to protect the host against CVB3-initiated VM.
-
HeLa and HEK-293 T cells were the gifts from Dr. Huaiqi Jing [National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention (Beijing, China)], and were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, 12430104; Thermo Fisher Scientific Inc., Waltham, MA, USA) with 8% fetal bovine serum (FBS; Gibco, 10099141C; Thermo Fisher Scientific Inc.) and 100 lg/mL penicillin–streptomycin (Gibco, 10378016; Thermo Fisher Scientific Inc.) at 37 ℃ in a humidified atmosphere with 5% CO2 (Wang et al. 2019). Pathogen-free 3-week-old BALB/c mice were obtained from the Jiangsu University Experimental Animal Center (Zhenjiang, China).
-
CVB3/Nancy (Corsten et al. 2015), a gift from Professor Ruizhen Chen (Department of Cardiology, Zhongshan Hospital, Shanghai, China), and EGFP-CVB3, which expresses the enhanced green fluorescence protein (EGFP), are constructed previously based on pMKS1, a plasmid containing the full-length CVB3 genomic complementary DNA (a gift from Professor Zhaohua Zhong, Harbin Medical University, Harbin, China) (Tong et al. 2013; Wu et al. 2014). Adeno-associated viruses (AAV) were expressed as Renilla luciferase (RLuc); HBAAV2/9-TNT-Null-RLuc was used as a negative control (Stein et al. 2015).
-
The 3-week-old BALB/c male mice were injected through the tail vein with AAV (11 mice per group). The mice were reared under standard laboratory conditions for the next four weeks. And after successful RLuc expression, CVB3 (105 PFU/mouse) was injected intraperitoneally. For injection, CVB3 was diluted in 100 μL PBS, and an equal volume of PBS was injected into the blank control mice (Yuan et al. 2009).
-
The miRNAs, including miR-324-3p (5′-CCCACUGCCC CAGGUGCUGCUGG-30), miR-324-3p inhibitor (5′-CCA GCAGCACCUGGGGCAGUGGG-3′), siRNA TRIM27 (si-TRIM27) (5′-GGCAGUGUCUUUGUGGUAUTT-3′), the miRNA negative control (miR-NC), and inhibitor NC were synthesized by GenePharma Co., Ltd. (Suzhou, China). Cells were seeded with a density of 2.5 × 105 cells/well (in a 6-well plate) in DMEM with 8% FBS. Lipofectamine 3000 (L3000015; Invitrogen, Carlsbad, CA, USA) and miRNA were mixed in 250 μL of DMEM at room temperature for 10 min, and then were added to the cells (Zhang et al. 2019).
-
Total cellular RNA was extracted by Trizol (T9424; Sigma-Aldrich, Saint Louis, MO, USA), and the non-coding small nuclear RNA (snRNA) U6 snRNA was detected as the endogenous control. RNA was reversed transcribed into cDNA using an RNA reverse transcription kit (RR036Q; TaKaRa Bio Inc., Shiga, Japan). The RT-qPCR analysis of VP1, GAPDH, and the other genes was performed using a RT-qPCR kit (RR830; TaKaRa Bio Inc.). All measurements were performed at least in triplicate (Wu et al. 2009; Mohamud et al. 2019).
-
CVB3supernatants were seriallydiluted and added into HeLa cells in 12-well plates (1 × 105 cells/well). After incubating the cells for 1 h, the cells were washed for three times with PBS, overlaid with 0.75% low melting point agarose (25 mL 1.5% agarose mixed with 25 mL 2 × DMEM), andincubated for 72 h. Cells were fixed with a fixative (25 mL acetic acid mixed with 75 mL absolute ethyl alcohol) for at least 30 min. After aspirating off the fixative, 1% crystal violet was added for30 min, andthecellswerewashedagainwithPBSforthree times (Garmaroudi et al. 2010).
-
Cells were lysed with RIPA buffer containing 1% phenylmethylsulfonyl fluoride (PMSF). Proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene fluoride (PVDF), and detected with the appropriate antirabbit primary antibodies (Amitava et al. 2011): GAPDH (ab8245; Abcam, Cambridge, UK), TRIM27 [D5S4O; Cell Signaling Technology, Inc. (CST), Danvers, MA, USA], ERK1/2 (#9102; CST), p-ERK1/2 (#4370; CST), and use a goat anti-rabbit IgG HRP-conjugated secondary antibody (128-005-160; Jackson ImmunoResearch, Inc., PA, USA). ERK1/2 inhibitor was purchased from MedChemExpress LLC., (MCE, PD98059, Monmouth Junction, NJ, USA).
-
The tissue samples were fixed in 10% (v/v) formalin (252549; Sigma-Aldrich), embedded in paraffin, and sectioned into 4 μm sections. Cross-sectioned tissue samples were stained with hematoxylin and eosin (H & E) (Wang et al. 2018).
To obtain a quantitative estimation of the histological cardiac damage, sections (n = 6 for each experimental group) were scored by two independent observers blinded to the experimental protocol. The following morphological criteria were used: score 0, no damage; score 1 (mild), interstitial edema and focal necrosis; score 2 (moderate), diffuse myocardial cell swelling and necrosis; score 3 (severe), necrosis with presence of contraction bands and neutrophil infiltrate; score 4 (highly severe), widespread necrosis with presence of contraction bands, neutrophil infiltrate, and hemorrhage.
-
The tissue samples were fixed in 10% (v/v) formalin, embedded in paraffin, and sectioned into 4 μm sections. Tissue slides were deparaffinized and rehydrated through an alcohol series, followed by antigen retrieval with sodium 0.01 mol/L citrate buffer (pH = 6.0) for antigen retrieval. Tissue was blocked with 5% bovine serum albumin (A1933; Sigma-Aldrich) with 0.1% Triton X-100 and 3% H2O2 in PBS for 1 h at room temperature and then incubated with appropriate primary antibodies at 4 ℃ overnight. Stained with DAB reagent and counterstained with hematoxylin, and observed under a light microscope.
-
Cells cultured in collagen-coated chamber slides (NEST Biotechnology Co., Ltd., Wuxi, China) were washed and fixed with either 4% paraformaldehyde or with ice-cold methanol. Cells were permeabilized with 0.1% Triton X-100 in PBS and incubated overnight with the indicated primary antibodies at 4 ℃. After washing, cells were incubated with the appropriate secondary antibody for 60 min at 37 ℃ (Ye et al. 2014).
-
To determine cytotoxicity, we measured the concentration of LDH in the cell-free supernatants according to the manufacturer's instructions (MK401; Clontech, Mountain View, CA, United States).
-
Statistical analysis was performed using the Graph Pad Prism 7.0 (GraphPad Software Inc., San Diego, CA, USA). All data are expressed as the mean ± standard deviation (X ± SD), and the difference was statistically significant when *P < 0.05, **P < 0.01, or ***P < 0.001.
Cell Culture and Animals
Virus
Myocarditis
Plasmid, miRNAs and siRNAs Transfection
RNA Extraction, RT-PCR and Quantitative PCR
Viral Plaque Assay
Western Blot Analysis
Histopathology
Immunohistochemistry (IHC) Staining
Immunofluorescence (IF) Microscopy
Lactate Dehydrogenase (LDH) Release Assay
Statistical Analysis
-
To investigate the roles of miR-324-3p in CVB3 infection, in particular, the association between viral infection and miR-324-3p expression levels, HeLa cells were infected with CVB3 and the miR-324-3p levels were determined. RT-qPCR analysis showed that the miR-324-3p expression level was increased in CVB3-infected HeLa cells (Fig. 1A). To determine the effect of miR-324-3p on CVB3 replication and transcription, RT-qPCR and Western blot analyses were used to measure the expression levels of viral genomic RNA and capsid protein VP1. The effects of miR-324-3p overexpression or silencing in CVB3-infected HeLa cells were also investigated. The RT-qPCR analysis results showed that, compared with the miR-NC, miR-324-3p significantly decreased VP1-coding RNA levels (Fig. 1B). Western blot analysis results revealed that, compared with miR-NC or inhibitor-NC, overexpression of miR-324-3p decreased VP1 expression, whereas silencing of miR-324-3p increased VP1 expression (Fig. 1C, 1D).
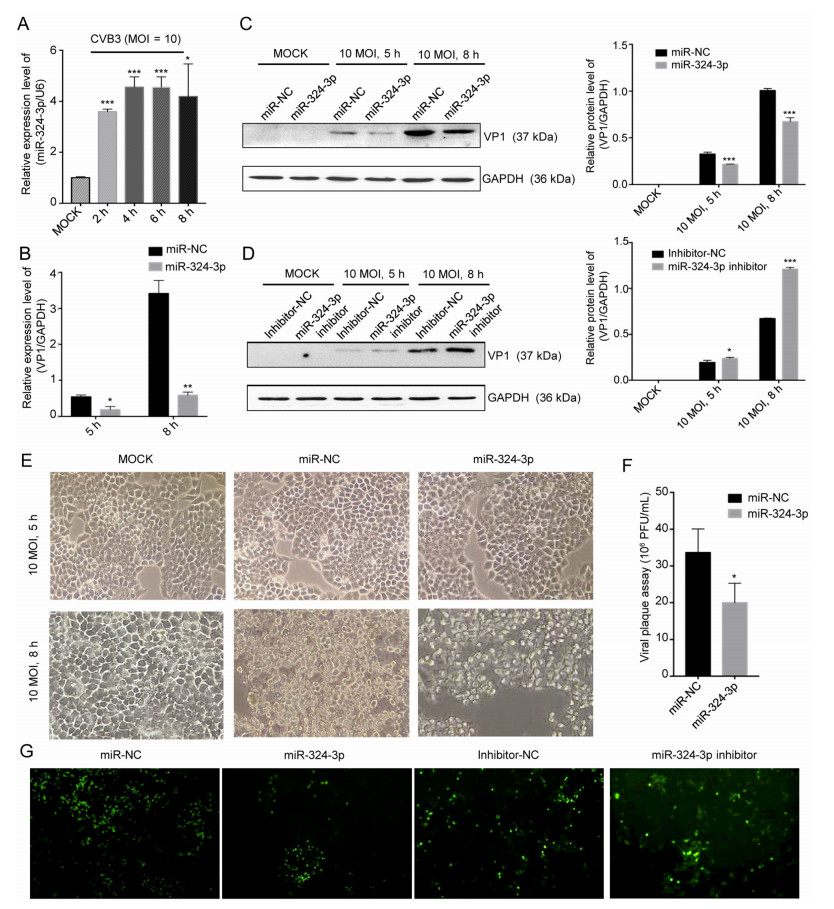
Figure 1. miR-324-3p suppresses CVB3 replication. A HeLa cells were infected with CVB3 (MOI = 10); RT-qPCR analysis was used to detect the expression level of miR-324-3p (n = 3). B HeLa cells were transfected with miRNA and cultured for 48 h, then infected with CVB3 (MOI = 10); total RNA and protein were extracted for the detection of VP1 by RT-qPCR analysis (n = 3). C, D HeLa cells were transfected with miRNA and cultured for 48 h, then infected with CVB3 (MOI = 10); total protein was extracted for the detection of VP1 by Western blot analysis. E Cytopathic effect (CPE) was observed. HeLa cells were transfected as described above and cell morphology was analyzed by microscopy after 5 h and 8 h of CVB3 infection (original magnification × 400). F Viral plaque assay. HeLa cells were transfected with miR-324-3p and miR-NC for 48 h, infected with CVB3 (MOI = 0.01) for 72 h (n = 3). G HEK-293 T cells were co-transfected with miRNA and EGFP-CVB3. A fluorescence microscope was used to observe EGFP-positive cell number after 48 h (original magnification × 100). CVB3, coxsackievirus B3; MOI, multiplicity of infection.
The inhibitory effect of miR-324-3p on CVB3 replication and biosynthesis was further verified by microscopy, which revealed that the cytopathic effect (CPE) on HeLa cells transfected with miR-324-3p was significantly suppressed compared with miR-NC (Fig. 1E). The plaque assay revealed that miR-324-3p significantly suppressed CVB3 replication (Fig. 1F). Additionally, fluorescence microscopy revealed that EGFP expression was lower in HEK-293 T cells transfected with miR-324-3p than in those transfected with miR-NC. Also, silencing of miR-324-3p, resulted in higher number of EGFP expressing cells compared with those transfected with inhibitor-NC (Fig. 1G).
VP1 was localized in both cytoplasm and nucleus, but its accumulation in nucleus was obvious. Other studies have shown that VP1 localized in nucleus can enhance CVB3 replication by influencing cell cycle. IF staining results showed that VP1 was scattered in cell, with fluorescence located in cytoplasm, but no significant difference was detected between the miR-324-3p and miR-NC groups in CVB3-infected cells at 5 h (Fig. 2A). In CVB3-infected cells at 8 h, the overexpression of miR-324-3p group showed less IF signal of VP1, especially in nucleus compared with miR-NC group. On the contrary, the VP1 IF signal in cells transfected the miR-324-3p inhibitor was more potent than inhibitor NC group when CVB3 infected HeLa cells in 5 h and 8 h (Fig. 2B).
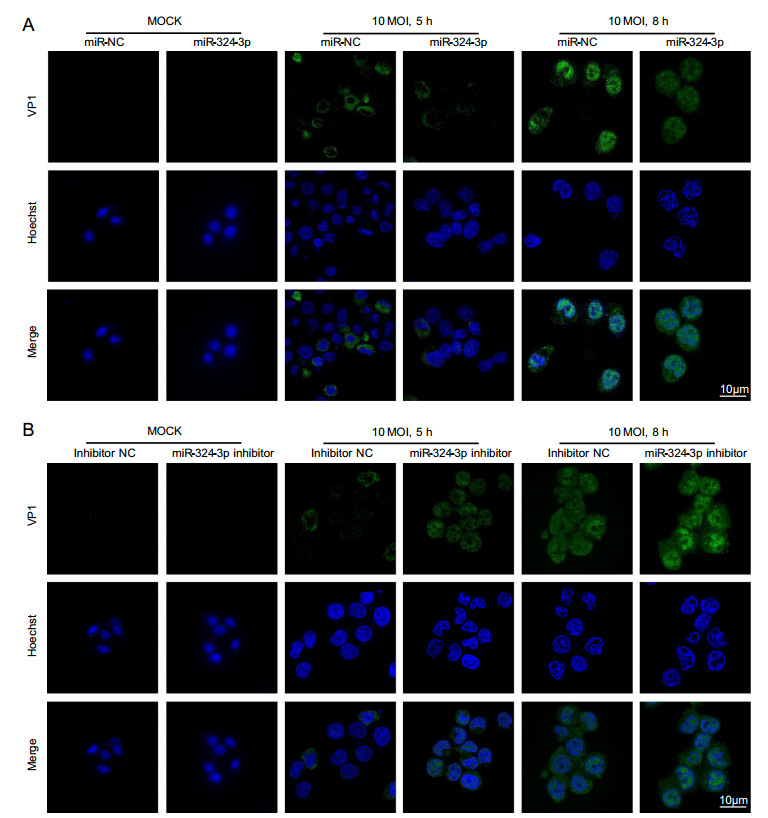
Figure 2. miR-324-3p influences the expression level of VP1. A IF microscopy analysis showed the expression of VP1 in HeLa cells after overexpressing miR-324-3p and infected with CVB3 (MOI = 10) for mock, 5 h and 8 h. B IF microscopy analysis showed the expression of VP1 in HeLa cells after transfected with miR-324-3p-inhibitor and infected with CVB3 (MOI = 10) for mock, 5 h and 8 h. IF, immunofluorescence; CVB3, coxsackievirus B3; MOI, multiplicity of infection; scale bar: 10 μm.
-
Using the platform TargetScan (http//www.targetscan.org/), we found a binding site of miR-324-3p at the 30-UTR region of trim27 from human tissues (Fig. 3A). According to the results of the dual-luciferase reporter gene assay in HEK-293 T cells, the luciferase activity of TRIM27-30UTR-mut did not show noticeable difference after transfection with miR-324-3p mimic and NC. However, compared with NC, the miR-324-3p mimic dramatically reduced the luciferase activity of TRIM27-30UTR-wt (Fig. 3B), suggesting that trim27 might be a direct target gene of miR-324-3p. The RT-qPCR and Western blot analysis results showed that miR-324-3p had a negative effect on TRIM27 expression (Fig. 3C, 3D).
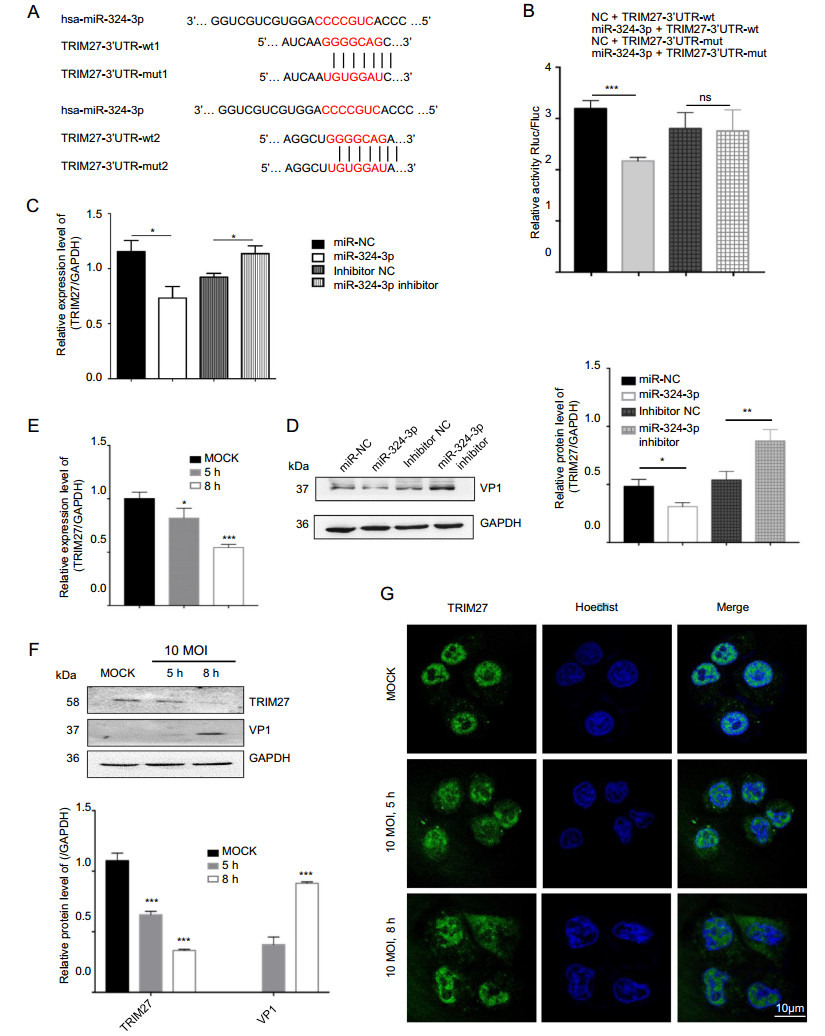
Figure 3. CVB3 infection and miR-324-3p regulate the expression level of TRIM27. A Prediction of miR-324-3p targeting sequence on trim27. B Validation of miR-324-3p targeting on trim27 by luciferase assay (n = 3). C, D HeLa cells were transfected with miRNA for 48 h; the expression level of TRIM27 was analyzed by Western blot and RT-qPCR (n = 3). E RT-qPCR analysis was done to measure the expression level of TRIM27 after HeLa cells infected with CVB3 (n = 3). F The protein level of TRIM27 was analyzed by Western blot after HeLa cells infected with CVB3 for 5 h and 8 h (MOI = 10). G The expression of TRIM27 in HeLa cells was analyzed by IF after infected with CVB3 for 5 h and 8 h (MOI = 10). CVB3, coxsackievirus B3; MOI, multiplicity of infection; IF, immunofluorescence; scale bar: 10 μm.
To determine the expression level of TRIM27 in HeLa cells after infected with CVB3, total RNA and protein were extracted to perform RT-qPCR and Western blot analyses (Fig. 3E, 3F). The data indicated that CVB3 infection decreased the expression level of TRIM27. IF microscopy analysis results showed TRIM27-positive staining in the nucleus of HeLa cells, but after CVB3 infection, besides the nucleus, it was clearly elevated in cytoplasm (Fig. 3G).
-
The ERK1/2 have an important role in virus replication in host cells. To confirm the role of TRIM27 in virus replication, we used the ERK1/2 inhibitor (PD98056) to inhibit ERK1/2 activation, infected cells with CVB3 and analyzed TRIM27 and VP1 expression level (Fig. 4A). The results showed that, PD98056 inhibited p-ERK1/2 activation and decreased VP1 and TRIM27 expression. HeLa cells transfected with empty plasmid vector (vector) or TRIM27 overexpression plasmid (TRIM27 vector), were treated with DMSO or PD98056, and were infected with CVB3 for 8 h. Western blot analysis was used to examine the relationship between TRIM27, CVB3 and ERK1/2 (Fig. 4B, 4C). The result showed that, without CVB3 infection, the ERK1/2 expression level had no influence on TRIM27, whereas with CVB3 infection, inhibition of ERK1/2 activation led to decreased TRIM27 expression.
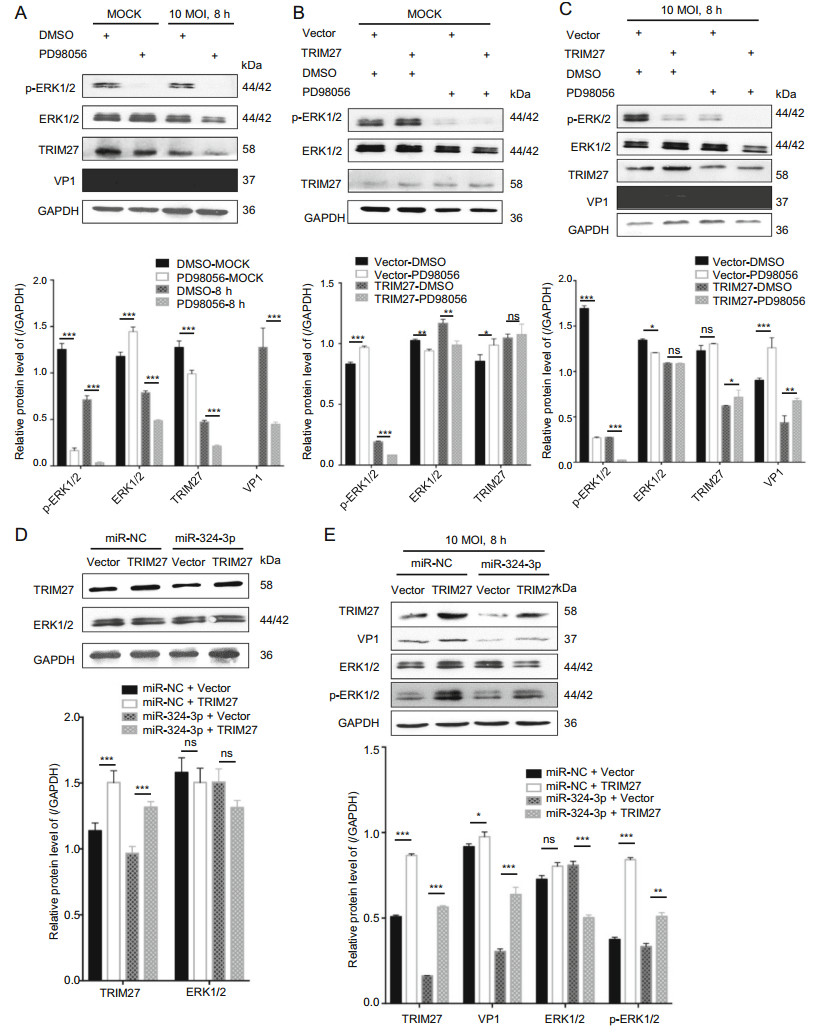
Figure 4. TRIM27 regulates CVB3 replication through ERK1/2 signaling pathway. A HeLa cells were treated with PD98056 or DMSO for 12 h, and infected with CVB3 (MOI = 10) for 8 h. Western blot analyzed the expression levels of VP1, ERK1/2, p-ERK1/2, and TRIM27, using GAPDH as control. B, C HeLa cells were transfected with empty plasmid vector or TRIM27 vector for 24 h, treated with PD98056 or DMSO for 12 h, and infected with CVB3 (MOI = 10) for 8 h. Western blot was applied to determine the expression level of VP1, ERK1/2, p-ERK1/2, and TRIM27, using GAPDH as control. D, E HeLa cells were co-transfected with miRNA mimics and empty plasmid vector or TRIM27 vector, and infected with CVB3. The expression levels of TRIM27, VP1, ERK1/2, and p-ERK1/2 were measured by Western blot analysis, using GAPDH as a loading control. CVB3, coxsackievirus B3; MOI, multiplicity of infection.
To further confirm TRIM27 downregulation by miR-324-3p-mediated inhibition of CVB3 replication, we overexpressed TRIM27 in the presence of miR-324-3p by co-transfection. The Western blot analysis data showed that HeLa cells overexpressed TRIM27 when transfected with the TRIM27 vector. And compared with cells cotransfected the miR-NC, co-transfected the miR-324-3p could inhibit TRIM27 expression. Additionally, without CVB3 infection, the expression levels of ERK1/2 were not significantly different between cells transfected with empty vector and TRIM27 vector (Fig. 4D).
After 8 h of CVB3 infection, miR-NC + TRIM27 vector increased the production of VP1 and p-ERK1/2 compared with miR-NC + empty plasmid vector (Fig. 4E). Co-transfection of the TRIM27 vector with miR-324-3p reduced the production of VP1, p-ERK1/2.
To further verify the upregulated effect of TRIM27 on CVB3 replication, we transfected HeLa cells with the TRIM27 vector to overexpress TRIM27 and infected them with CVB3. Western blot analysis data showed that overexpression of TRIM27 upregulated the expression level of VP1 and p-ERK1/2 in CVB3-infected cells at 8 h (Fig. 5A). Furthermore, these findings were further confirmed by plaque assay, which showed that overexpression of TRIM27 resulted in a significant increase of CVB3 progeny release (Fig. 5C). In contrast, dramatic suppression of TRIM27 expression by silencing TRIM27 using specific siRNA in CVB3-infected HeLa cells resulted in suppressed production of VP1, while p-ERK1/2 expression levels were decreased (Fig. 5B).
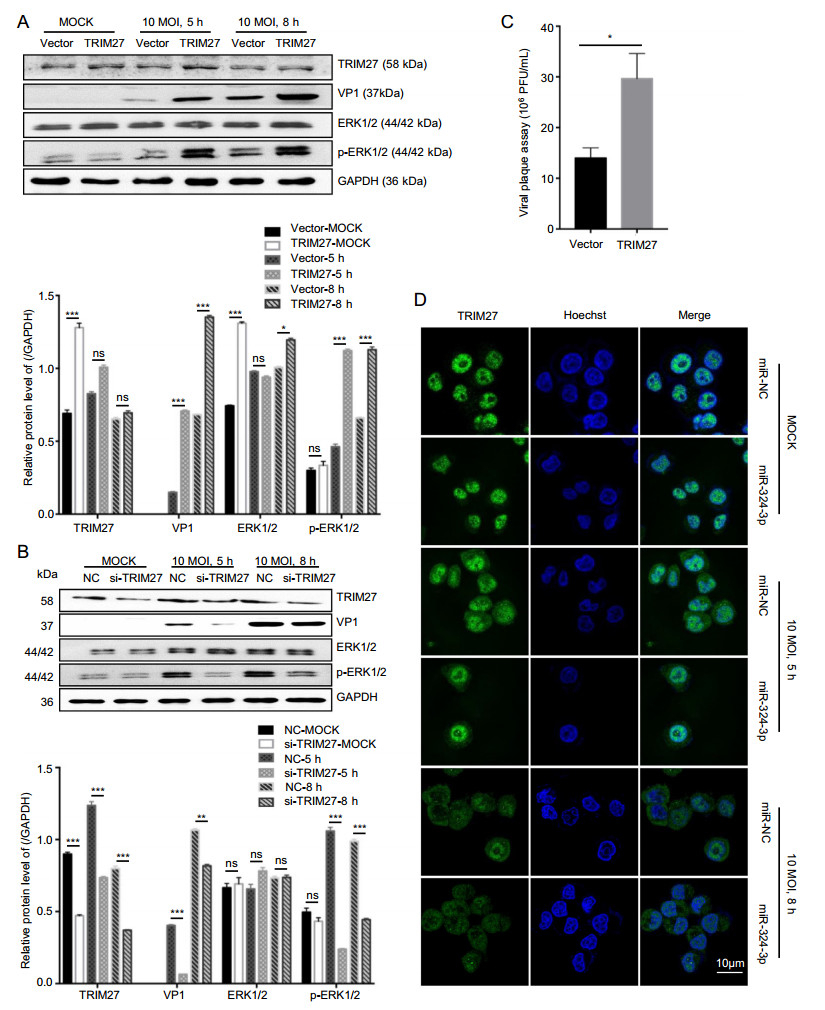
Figure 5. The role of TRIM27 in CVB3 replication. A, B HeLa cells were transfected with TRIM27 vector or si-TRIM27, and infected with CVB3 (MOI = 10) for 5 h and 8 h; total protein was extracted to analyze the expression level of VP1, TRIM27, ERK1/2 and p-ERK1/ 2, using GAPDH as a loading control. C Viral plaque assay. HeLa cells were transfected with miR-324-3p and miR-NC for 48 h, and infected with CVB3 (MOI = 0.01) for 72 h (n = 3). D HeLa cells were transfected with miRNAs and infected with CVB3 (MOI = 10) for 8 h. IF microscopy analyzed the expression of TRIM27. CVB3, coxsackievirus B3; MOI, multiplicity of infection, scale bar: 10 μm.
In our study, the expression level of TRIM27 was decreased after infected with CVB3. Specifically, the expression of TRIM27 in CVB3-infected cells decreased in nucleus but increased in cytoplasm. Overexpression of miR-324-3p in HeLa cells downregulated the TRIM27 expression level (Fig. 5D).
-
To investigate the role of miR-324-3p in CVB3-induced VM in vivo, we overexpressed or silenced miR-324-3p by AAV (or their negative control) in the mouse model. After the mice infected with CVB3 to establish the VM model, the miR-324-3p expression profile in mouse heart tissues was detected by RT-qPCR analysis. The results showed that miR-324-3p was successfully overexpressed or silenced in mouse heart tissues. In particular, the miR-324-3p expression level was increased in the VM mice compared with the control mice (Fig. 6A). The comparison of the body weight of the mice between different groups showed that VM led to the decreased body weight. The results also revealed that the mice overexpressing miR-324-3p were heavier than those in the control group, but lighter in the silencing group (Fig. 6B).
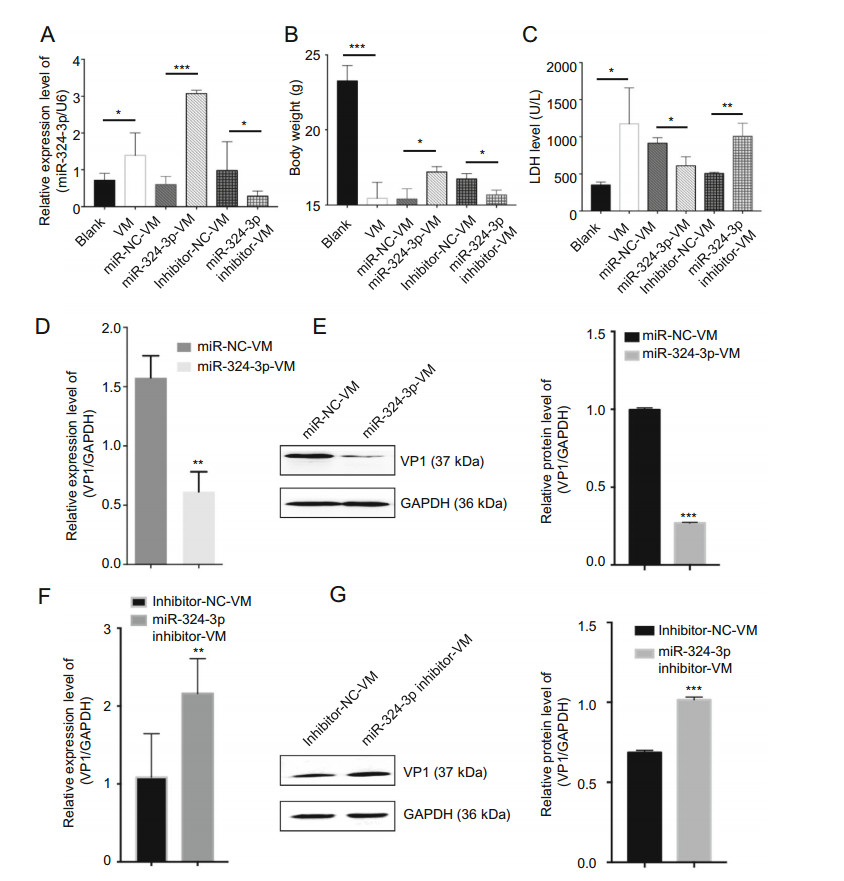
Figure 6. The function of miR-324-3p on CVB3-induced VM. A RTqPCR was employed to test the expression level of miR-324-3p in mouse heart tissues (n = 3). B The body weight of mice in different groups were recorded (n = 3). C Mouse serum was extracted to detect the level of lactate dehydrogenase (LDH) (n = 3). D, F The expression level of VP1-coding RNA was measured by RT-qPCR (n = 3). E, G The protein level of VP1 was detected by Western blot analysis (n = 3). VM, viral myocarditis.
Since LDH is abundantly distributed in myocardium with obvious specificity, it can be used for the diagnosis of myocardial diseases. The LDH analysis data indicated that LDH was significantly elevated in VM mice. The mice overexpressing miR-324-3p had lower LDH expression levels than that of the control group, whereas in mice of the miR-324-3p-inhibitor group, LDH was highly expressed compared to its control group (Fig. 6C). In addition, the evaluation of CVB3 replication by analyzing the expression of VP1 in mouse heart tissues revealed that, overexpression of miR-324-3p markedly suppressed viral replication (Fig. 6D, 6E), while knockdown of miR-324-3p significantly exacerbated viral replication, compared with their respective control (Fig. 6F, 6G).
In conclusion, CVB3-infected mice showed increased expression level of miR-324-3p. miR-324-3p negatively regulated CVB3 replication and viral load in mouse heart tissues.
-
The role of miR-324-3p regulation of TRIM27 expression was investigated in vitro in cell lines and in vivo in mouse heart tissues by RT-qPCR, Western blot and IHC analyses. Overexpression of miR-324-3p led to decreased TRIM27 expression level, whereas inhibition of miR-324-3p expression increased TRIM27 expression level (Fig. 7A). Western blot analysis results showed that the overexpression of miR-324-3p in VM mouse suppressed the protein expression levels of TRIM27, VP1 and p-ERK1/2 (Fig. 7B). TRIM27 protein was mainly expressed in cytoplasm, appearing with a brownish-yellow color. Compared with the blank control group, TRIM27 expression decreased in VM mice (Fig. 7C). Noteworthy, miR-324-3p reduced TRIM27 expression in cardiomyocytes of the mice in both CVB3-negative and VM groups.
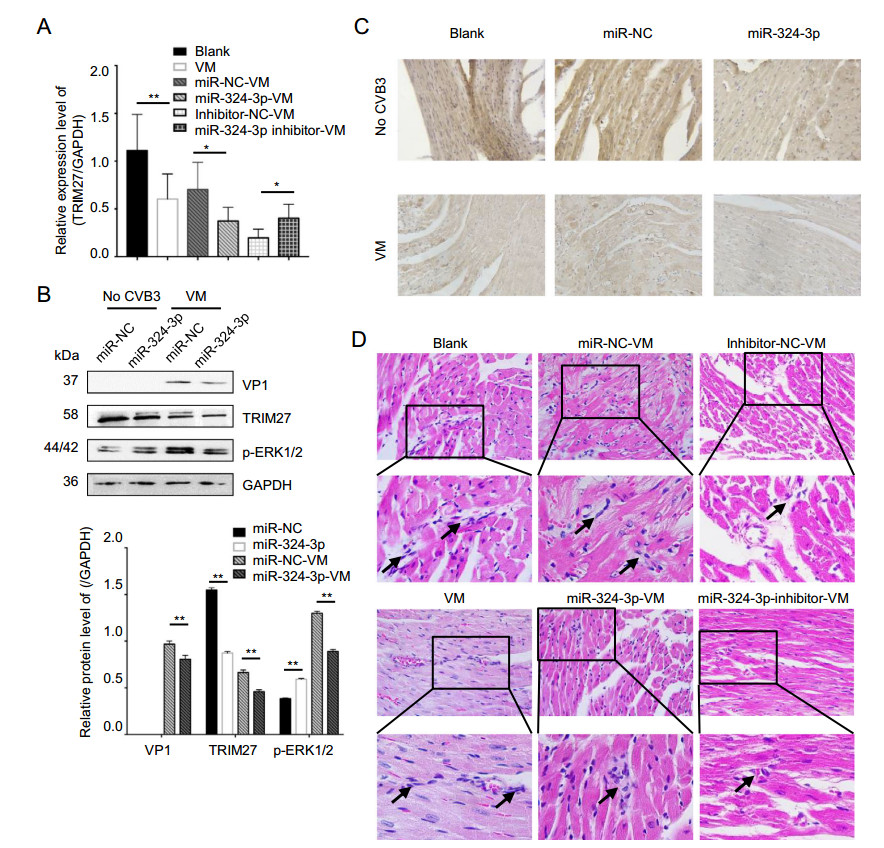
Figure 7. The role of miR-324-3p in VM. miR-324-3p was overexpressed or silenced by AAV injection in mice for 4 weeks. The mice were then infected with CVB3 (105 PFU/mouse) for next 3 weeks. Samples were collected for analysis. A RT-qPCR analysis was used to determine the expression of TRIM27 (n = 3). B Western blot analysis was used to determine the protein levels of VP1, TRIM27 and p-ERK1/2. GAPDH was used as a loading control. C The expression level of TRIM27 in mouse heart tissue was measured by immunohistochemical (IHC) analysis (original magnification × 400). D Heart tissue sections were prepared 3 weeks post-infection (n = 3). Histopathological evaluation was performed to detect the presence and severity of myocarditis in the heart tissues of different groups (original magnification × 400). The partial enlarged image enlarges 4.125 times of the original image, and the arrow points out where the infiltrating white blood cells. VM, viral myocarditis; AAV, adenoassociated viruses; CVB3, coxsackievirus B3.
Cardiac histopathology revealed that CVB3-infected mice overexpressing miR-324-3p showed only minor edema and hemorrhage, as well as inflammatory cell infiltration, while mice in miR-NC group showed significant necrosis and signs of mononuclear cell infiltration. Furthermore, more scattered small foci in the cardiac tissues of CVB3-treated mice were observed in the group inhibiting miR-324-3p. In addition, more cardiac infiltration of inflammatory cells and necrosis of myocardial cells, as well as the destruction of myocardial structure, were observed in CVB3-infected mice. The score of VM, miRNC-VM, miR-324-3p-VM, inhibitor-NC-VM and inhibitor-324-3p-VM groups were 3.083 ± 0.229, 2.750 ± 0.218, 1.833 ± 0.207, and 2.417 ± 0.193, respectively (Fig. 7D).
CVB3 Infection Increases the Expression of miR-324-3p and miR-324-3p Inhibits CVB3 Replication
Trim27 Is A Target Gene of miR-324-3p
miR-324-3p Regulates CVB3 Infection and Replication by Targeting TRIM27
miR-324-3p Reduces Mice Viral Myocarditis Symptom
miR-324-3p Targets TRIM27 to Alleviate CVB3-Induced Inflammation and Myocarditis
-
VM refers to the inflammation of the heart muscle that can be initiated by a variety of viral infections (Maisch et al. 2000). Clinically, adult patients always present as highly variable acute myocarditis, ranging from subclinical disease to fulminant heart failure. The severity of symptoms is dependent on age. Newborns and infants are often more severely affected (Blauwet and Cooper 2009). In adults, chronic forms occur more often. Only a small percentage of adult patients exert fulminant myocarditis (Martin et al. 1994; McCarthy et al. 2000). CVB3 is thought to be a major etiologic agent of VM. However, despite extensive research, the pathogenesis of CVB3-induced myocarditis is still unclear, and its investigation and therapy options are limited (Van Linthout et al. 2014). miRNAs are small noncoding RNAs whose regulatory effects exert either through mRNA destabilization or translation inhibition by binding to complementary sequences within the 30-UTR of their target mRNAs. In the past few years, miRNAs have been found to have key roles in infectious and inflammatory diseases (Mehta and Baltimore 2016). In this study, we investigated the effects of miR-324-3p on the viral myocardial inflammation induced by CVB3, as well as their underlying mechanisms. The study showed that, miR-324-3p not only reduced CVB3 replication in vitro, but also played a negative regulatory role in CVB3-induced myocardial inflammation in vivo.
Viruses are cellular parasites that replicate and assemble either in the cytoplasm or the nucleus (Schang 2003). CVB3 is no exception. The genome of CVB3 encodes four structural proteins (VP1, VP2, VP3, and VP4) (Bowles et al. 1986). The viral capsid protein, VP1, controls the cell cycle of the host cell, by promoting the successful expression of viral proteins, thereby leading to enhanced production of viral progenies, and also seems to play a key role in the life-cycle of CVB3 (Wang et al. 2017). In previous studies, the VP1 of CVB3-infected cells was found in the nucleus of the cells, which produced more viral protein in their G1 and G1-S phase (Amy and Skotheim 2013). A shortening of G2, as confirmed in other study, might lead to a higher production of viral proteins by the host cell, resulting in higher production of CVB3 progeny (Feuer et al. 2004; Feuer and Whitton 2008). In earlier studies, VP1 was reported to have a potential effect on CVB3 transport and nuclear localization. In our research, miR-324-3p reduced CVB3 replication and biosynthesis, attenuating CVB3-induced CPE in HeLa cells and cardiomyocyte injury in mice. The Western blot and RT-qPCR results showed that miR-324-3p decreased CVB3 VP1 expression. IF staining results revealed that miR-324-3p inhibited VP1 expression in HeLa cells and at the same time decreased VP1 localization in the nucleus. One of the reasons for the development of CVB3-induced VM is the direct damage to cardiomyocytes caused by CVB3 replication, which leads to CPE in cells and disturbs the cell cycle. The results also suggest that the mechanism for the mitigation of CVB3-induced VM by miR-324-3p involves not only the reduction of CVB3 replication, but also the inhibition of the VP1 localization in the nucleus.
miR-324-3p plays a negative role in CVB3 replication, and has a negative regulatory effect on TRIM27 expression. IF staining analysis data showed that, TRIM27 was localized in the nucleus when HeLa cells lied in a resting state (during CVB3 infection). After the cells infected with CVB3, with the prolongation of infection, TRIM27 is translocated from the nucleus to the cytoplasm. The different localizations of TRIM27 have different effects on the function of cells. For instance, cell proliferation occurs when TRIM27 is localized in the cytoplasm, whereas when the TRIM27 protein is mainly localized in the nuclei of mesangial cells, lower proliferation level is observed.
In this study, we identified miR-324-3p as a regulator of CVB3 replication. It targets TRIM27 to exert its effects on the ERK1/2 pathway. In CVB3 infection, miR-324-3p reduces viral replication through ERK1/2 pathway, thereby attenuating CVB3-induced VM by decreasing the destruction of myocardial tissues. miR-324-3p reduces the localization of VP1 in the nucleus, and inhibits the CVB3-induced interruption of the cell cycle. To the best of our knowledge, this is the first report on the influence of miRNA on VP1 localization. These findings improve our understanding of the functional roles of miRNA in viral replication.
In conclusion, miR-324-3p alleviates CVB3-induced VM by targeting TRIM27.
-
The research was support by National Natural Science Foundation of China, Grant No. 81971945 and No. 81802013 (https://isisn.nsfc.gov.cn/egrantweb/), Xuzhou Science and Technology Project, Grant No. KC1717 (http://kjj.xz.gov.cn), the Projects from Social development of Zhenjiang, Grant No. SH2019044 (http://kjj.zhenjiang.gov.cn).
-
HXS, SHS and HW conceived and designed the experiments. TJL, JYL, YS, JT and YL performed the experiments. TJL, HXS and HW analyzed the data. TJL, HXS, SHS, CS, HW and JYQ contributed reagents/materials/analysis tools. TJL, HXS and HW wrote the paper.
-
The authors declare that they have no conflict of interest.
-
This study was conducted in strict accordance with the recommendations in the Guide to the Care and Use of Experimental Animals-Chinese Council on Animal Care. All protocols were approved by the Animal Care Committee of University Jiangsu, (protocol number: UJS-IACUC-AP-20190307087).


 DownLoad:
DownLoad: