









HTML
-
Viroplankton are the most abundant and diverse group of plankton existing in aquatic environments, often exceeding bacterial abundance by 10–100-fold(Bergh et al., 1989; Wommack and Colwell, 2000). Viruses are thought to infect up to 50%–100% of phytoplankton(Proctor and Fuhrman, 1990; Nagasaki et al., 2004), and 10%–66% of the bacterial community is lysed by viruses each day(Suttle, 1994; Baudoux et al., 2006). The substantial impact of viruses on bacterial, archaeal, and eukaryotic cell mortality suggests that aquatic environments are viral reservoirs of the greatest genetic diversity on Earth(Fuhrman, 1999; Wommack and Colwell, 2000; Suttle, 2005; Suttle, 2007).
Phytoplankton viruses are normally referred to as cyanophages and phycodnaviruses. Most of the characterized cyanophages, which infect cyanobacteria, belong to three families: Myoviridae, Podoviridae, and Siphoviridae(Suttle, 2000; Mann, 2003; Sabehi et al., 2012). Members of Phycodnaviridae infect a variety of eukaryotic microalgae. Using cell cultural methods, over 60 phytoplankton viruses have been isolated from aquatic environments, and their full-length genomic sequences have been determined(Sullivan et al., 2005; Fitzgerald et al., 2010; Sullivan et al., 2010). However, most of these viruses were cultivated from marine environments(Suttle, 2000; Dunigan et al., 2006), and few freshwater viral genomes have been identified. Indeed, the genomes of only three cyanomyoviruses, four cyanopodoviruses, one unclassified cyanophage, and seven phycodnaviruses have been reported from freshwater sources to date(Fitzgerald et al., 2007; Yoshida et al., 2008; Gao et al., 2012; Zhou et al., 2013). Within the last few decades, scientists have found that viruses are as prevalent in freshwater environments, including rivers(Farnell, 2003), alkaline lakes(Jiang et al., 2004), eutrophic lakes(Weinbauer et al., 2003), mesotrophic lakes(Wilhelm and Smith, 2000), and even oligotrophic lakes(Vrede et al., 2003), as they are in marine environments. However, the mechanisms for controlling viral production and loss are thought to be fundamentally different between marine and freshwater environments, as shown in a comparison of the relationships between viral and bacterial abundance(Clasen et al., 2008).
Understanding the biodiversity and population dynamics of viral communities in natural ecosystems is important. However, given the inherent difficulties of virus isolation from aquatic environments, these features are largely underestimated. In recent years, viral diversity has been explored using culture-independent techniques. In contrast, analysis of the diversity of aquatic viruses has been proven difficult because of the general lack of sequence conservation among viral genomes. In many studies of viral genomics, several genes have been found to be conserved among certain algae viruses, and appropriate genetic markers for elucidation of viral biodiversity have been established. Genetic markers, such as genes encoding photosynthesis reaction components(i.e., psbA), capsid proteins(mainly g20 and mcp), and DNA polymerase, have been widely used to explore the diversity and evolutionary history of phytoplankton viruses in marine and freshwater environments(Larsen et al., 2008; Labonte et al., 2009; Gimenes et al., 2012; Zheng et al., 2013; Zhong and Jacquet, 2013).
East Lake is a typically urban, shallow lake located in Wuhan, China. With a water area of over 30 km2, East Lake is a source of drinking water and aquaculture resources and also used for recreation and large-scale fishing. Over the last few decades, East Lake has frequently suffered from massive algae blooming because of eutrophication in water systems. Thus, it is likely that this lake harbors a diverse and dynamic community of viruses. Liu and colleagues(2006)assessed the spatial distribution and morphologic diversity of viroplankton in East Lake using transmission electron microscopy. They estimated that the lake contains 108–109 viral particles/mL water, with over 70 morphotypes observed. In addition, one virus(named PaV-LD)infecting the harmful filamentous cyanobacterium Planktothrix agardhii was isolated from this lake(Gao et al., 2012). Our previous study, which used a high-throughput sequencing technique, also confirmed that this lake contains a large number of viruses(Ge et al., 2013).
In the present study, we conducted a 1-year survey of phytoplankton viruses in East Lake using seven primers sets targeting conserved genes and found remarkably diverse viral sequences related to different phytoplankton virus families.
-
Water samples were collected monthly between May 2012 and April 2013 from four sites along East Lake in Wuhan, China. The environmental characteristics of the sampling sites are described in our previous report(Xia et al., 2013).
To quantify virus concentration, 100-L water samples were collected from the surface to a depth of 0.5 m at the four sites. Water samples were then mixed, transported to the laboratory in polyethylene bottles, and treated within 2 h, as previously described(Thurber et al., 2009). In brief, to remove zooplankton, phytoplankton, and large particles, the sample was serially prefiltered through 16 layers of gauze and a plankton net(200 μm mesh size). To remove bacteria, the preliminarily filtered water was then filtered with 1-and 0.45-μm nominal pore size polycarbonate filters using a peristaltic pump. The filtrate was concentrated approximately 50–100-fold to a final volume of 200–250 mL using a 10 kDa-cutoff tangential-flow ultrafiltration system(Millipore, USA). Particles of less than 0.45 μm were further concentrated by ultrafiltration at 111, 000 × g for 2 h at 4℃(Beckman Coulter Optima, SW-28, USA) and resuspended with 100–200 μL precooled phosphate buffer saline. The prepared viral concentrates( < 0.45 μm)were stored immediately in the dark at −80 ℃ until further processing.
-
Primer sets targeting the genes encoding minor capsid assembly proteins(g20)of cyanomyoviruses(Sullivan et al., 2008), DNA polymerases(polAL)of cyanopodoviruses(Chen et al., 2009), and DNA polymerases(polB)or major capsid proteins(mcp)of phycodnaviruses(Chen and Suttle, 1995; Larsen et al., 2008)were used in this study. No conserved gene has been found for the five known cyanosiphovirus genomes. Based on published data and sequences obtained from our previous metagenomic data(unpublished), three degenerate primer sets separately targeting ribonucleotide reductase genes(RNR), large terminase subunit genes(terL), and major capsid protein genes(mcp)of cyanosiphoviruses were designed(Table 1). These three primer sets covered the three subtypes of cyanosiphovirus, i.e., S-CBS4, S-CBS2/PSS2, and S-CBS1/S-CBS3, respectively(Huang et al., 2012).
Gene Virus Primer name Sequence (5'-3') TM1/TM2 (℃) References g20 Cyanomyovirus CPS1.1 F
CPS8.8 RGTAGWATWTTYTAYATTGAYGTWGG
ARTAYTTDCCDAYRWAWGGWTC35/46 (Sullivan et al., 2008) polA Cyanopodovirus CP-DNAP-349F
CP-DNAP-533Ra
CP-DNAP-533RbCCAAAYCTYGCMCARGT
CTCGTCRTGSACRAASGC
CTCGTCRTGDATRAASGC48/58 (Chen et al., 2009) RNR S-CBS4 subtype RNR F
RNR RATGGMATYGAAGCVWSYTGGCG
AAYGTHTGCYTGGARGTWTAYCT43/55 This study terL S-CBS1/S-CBS3 subtype TerL F
TerL RCGTGTKGTGCTGATGTT
TCARCGCHGAKGGCYTGAT37/50 This study mcp S-CBS2/PSS2 subtype MCP F
MCP RTGSCCGRMGTkGSYTTCCGT
CCAYTCRAbGCGRGTSA42/56 This study mcp Phycodnavirus mcp F
mcp RGGYGGYCARCGYATTGA
TGIARYTGYTCRAYIAGGTA45/58 (Larsen et al., 2008) polB Phycodnavirus AVS1
AVS2
PolGARGGIGCIACIGTIYTIGAYGC
GCIGCRTAICKYTTYTTISWRTA
SWRTCIGTRTCICCRTA40/52 (Chen and Suttle, 1995) Note: g20: minor capsid assembly protein genes of cyanomyoviruses; polA: DNA polymerase I genes of cyanopodoviruses; RNR, terL, and mcp: ribonucleotide reductase genes, large terminase subunit genes, and major capsid protein genes of cyanosiphoviruses, respectively; polB and mcp: DNA polymerase genes of family B and major capsid protein genes of phy-codnavirus, respectively Table 1. Primer sets used for amplification of algal virus-like sequences from East Lake.
-
Viral concentrates were digested with 0.1 U DNase(Promega, China) and 0.1 μg RNase(Fermentas, LTU)to remove host nucleic acid, according to our previously reported protocol(Xia et al., 2013). Fifty microliters of viral DNA extracted from a 140-μL sample using the QIAamp MinElute Virus Spin Kit(Qiagen, Germany)was used for detection. PCR was performed in a 50-μL reaction mix containing 5–10 ng nucleic acid for each sample, 25 × Mix Buffer, and 10 pmol of each primer(Beijing CoWin Bioscience Co., Ltd., China). Negative controls were set up using 1 μL of nuclease-free water as a template. A touch-up PCR program with a lower annealing temperature was used to improve the amplification efficiency. The PCR amplifications were carried out with a thermocycler(Bio-Rad, Hercules, CA, USA)with PCR programs as follows: denaturing at 95 ℃ for 5 min, followed by 45 cycles of denaturing at 95 ℃ for 30 s, annealing for 45 s, and extension at 72 ℃ for 1 min, with a final extension at 72 ℃ for 10 min. Different annealing temperatures were used for different primer sets. The annealing temperature was ramped from TM1 by 0.5 ℃ every cycle for first 20 cycles, followed by 25 cycles at constant annealing temperature TM2(Table 1). Each reaction was optimized to produce positive clones from the East Lake samples. The PCR products were analyzed by electrophoresis onto 1.5% agarose gels and stained with ethidium bromide. For sequencing, PCR products were excised from the agarose gel, purified with Gel Extraction Kits(OMEGA EZNA, Omega Bio-tek, USA), and cloned to T-vectors using the pGEM-T Easy System(Promega, Madison, WI, USA)following the manufacturer's instructions. About 10–15 r and om positive clones from each PCR product with expected sizes were sequenced with a universal primer set(M13)at Sangon Biotech(Shanghai, China).
-
The sequences were analyzed using BLAST at the NCBI website(http://blast.ncbi.nlm.nih.gov/Blast.cgi). All sequences were deposited in GenBank with accession numbers: KP775010–KP775086, KP775191–KP775342, and KP775408–KP775536. Multiple sequence alignments of amino acid consensus sequences were carried out using ClustalW and the Gonnet protein weight matrix.
Amino acid sequences from the same clone library with identities over 99% were removed before further analysis. Phylogenetic reconstruction was conducted using molecular evolutionary genetic analysis software(MEGA 5.0)(Kumar et al., 2008)with the neighbor joining(NJ)algorithmic method with 1000-fold bootstrap support and maximum likelihood(ML)phylogeny with 100 bootstrap replicates. Both NJ and ML methods for the seven genes were determined by the Jones-Taylor-Thornton(JTT)model and gamma-distributed rate heterogeneity among amino acid substitution(Zhong and Jacquet, 2013).
Collection and processing of water samples
Polymerase chain reaction(PCR)primers for phytoplankton viruses
Nucleic acid isolation, PCR amplification, and sequencing
Phylogenetic analysis
-
In this study, we obtained 358 different sequences that were homologous to phytoplankton viruses of four families. These sequences had similarities to known sequences of 65%–99% at the nucleotide level and 40%–99% at the amino acid level. Among these sequences, 77, 52, 100, and 129 were related to cyanomyoviruses, cyanopodoviruses, cyanosiphoviruses, and phycodnaviruses, respectively. Three virus families were detected in all 12 samples, with cyanopodovirus only detected in eight of 12 samples. Higher diversity was observed in cyanomyoviruses obtained in June, July, September, and November; cyanopodoviruses obtained in April, August, and November; and cyanosiphoviruses obtained from June to December. In contrast, the diversity of phycodnaviruses did not differ significantly throughout the year(Figure 1).
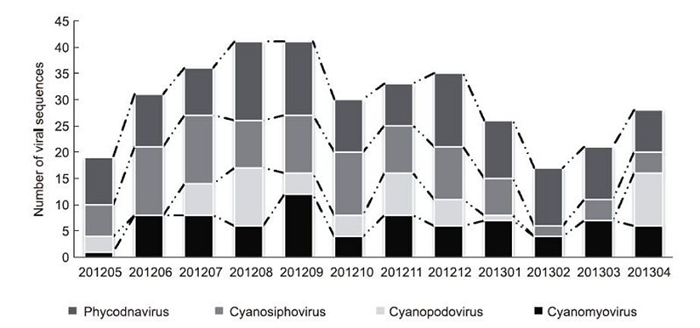
Figure 1. Temporal changes in viral abundance of different phytoplankton virus-like sequences, including cyanomyoviruses, cyanopodoviruses, cyanosiphoviruses, and phycodnaviruses. Viral populations were defined from the numbers of obtained viral sequences related to the seven genetic markers versus sampling months.
-
We identified 92 sequences as g20 homologs, varying from 501 to 601 bp in length. Similarity analysis revealed these g20 clones shared 54%–100% identity at the nucleotide level and 50.6%–100% similarity at the amino acid level. Translated protein sequences exhibited over 60% similarity with their closest relatives in the GenBank nr database. Among these sequences, 20 clones had over 90% amino acid identities with g20 fragments from freshwater or marine environments. A phylogenetic tree was constructed with 79 nonredundant g20 amino acid sequences from this study and representative g20 fragments from cyanophage isolates and environmental sequences(Figure 2). Five clusters(clusters α–ζ)were resolved in this tree, and all East Lake g20 sequences fell into these five clusters, forming distinct smaller groups(EL 1–5). Among these East Lake clones, 14, 24, 20, 24, and seven clones fell into clusters α, β, γ, δ, and ζ, respectively. Cluster α included only freshwater environmental clone sequences. Clusters β and γ included environmental clone sequences from both freshwater and marine water. In addition to environmental sequences, cluster δ included cyanophage isolates infecting Synechococcus or Prochloroccus. Four of the clusters agreed with previously published results(Wang et al., 2010), with the exception of cluster ζ, a novel freshwater cluster resolved in this study and formed by seven East Lake sequences, and one clone from the Kranji reservoir in Singapore.
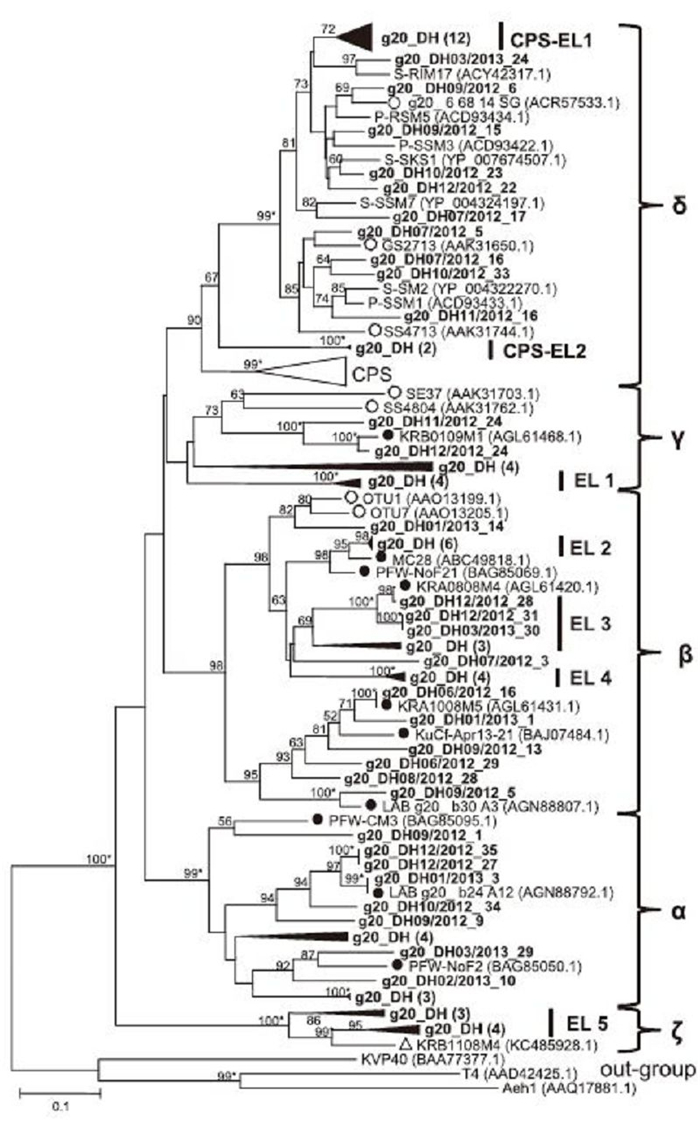
Figure 2. Neighbor-joining phylogenetic analysis of g20-related sequences from East Lake. Partial amino acid sequences of 79 g20-related genes from East Lake were aligned with those from other freshwater and marine environments and used for construction of a phylogenetic tree. CPS: a cluster containing g20 sequences from cultured Synechococcus phages. EL: groups containing only East Lake sequences (bold) with bootstrap values over 60%. Black triangles indicate groups of East Lake sequences with high similarity; the number of sequences is given in brackets. Filled and empty circles indicate published environmental sequences obtained from freshwater and marine systems, respectively. Numbers at tree branches indicate bootstrap values (> 50%) from 1000 replicates. Bootstrap values over 99% are labeled with *, and those that were less than 50% are not shown. The scale bar represents the number of amino acid substitutions per residue.
-
We identified 61 clones as polAL homologs, with sizes ranging from 568 to 589 bp. No polAL clones were detected in the June, February, and March samples. Similarity analysis showed that these sequences shared 33.6%– 100% nucleotide identity and 44.9%–100% amino acid identity. Their protein sequences had the highest similarity(50%–89%)with cyanopodoviruses or sequences found from Bay, Maine, or estuarine environments(Chen et al., 2009; Huang et al., 2010; Schmidt et al., 2014). Phylogenetic analysis was conducted with 52 different polAL amino acid sequences from this study and close relatives retrieved from GenBank(Figure 3). All clones fell into the largest cluster of marine picocyanobacteria podoviruses(MPPs)(Chen et al., 2009), except clone polA_DH2013/01_17, which was related to a bacterium phage. In this cluster, the subcluster MPP-B was formed, which included environmental sequences and five cultured podoviruses infecting Synechococcus or Prochloroccus. Within this subcluster, East Lake sequences formed at least three small independent groups(EL 1, EL 2, and EL 3): the largest group, EL 1, contained 34 sequences and was located in the same branch as cyanopodovirus S-CBP1; the medium group, EL 2, contained 12 sequences and was closely related to cyanopodovirus S-CBP4(Huang et al., 2010); and the smallest group, EL 3, contained only two sequences and was distantly related to the EL 1 and EL 2 groups.
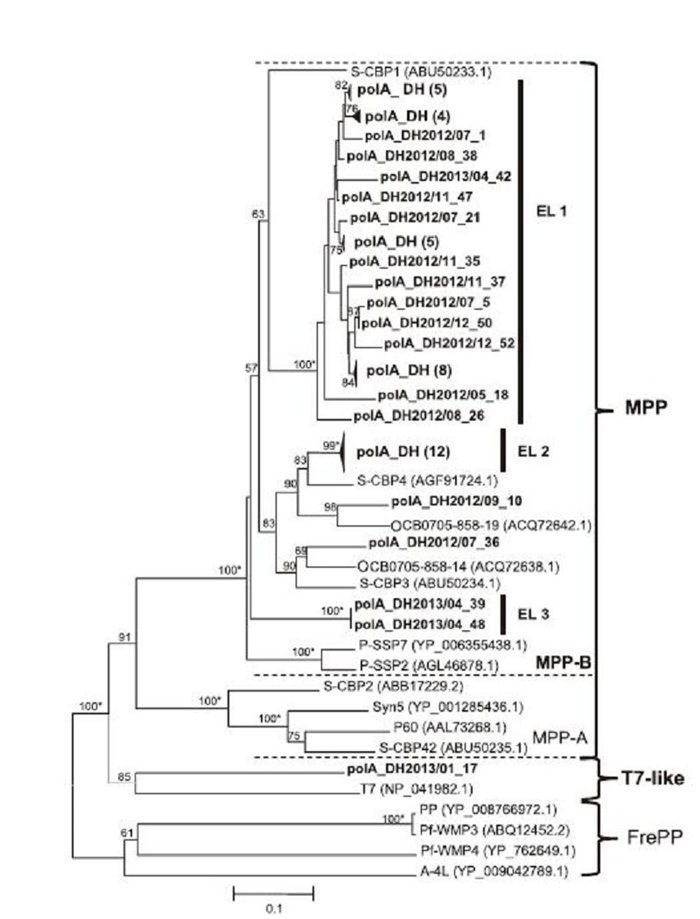
Figure 3. Neighbor-joining phylogenetic analysis of polA amino acid sequences from East Lake. Partial amino acid sequences of 52 polA-related genes from East Lake were aligned with sequences from cultured cyanopodovirus isolates and marine environments and used for construction of a phylogenetic tree. MPP: a cluster of polA sequences from cyanopodovirus isolates. EL: groups containing only East Lake sequences (bold) with bootstrap values over 99%. FrePP: a cluster of polA sequences of cyanopodoviruses from freshwater systems. Black triangles indicate groups of East Lake sequences with high similarity, with numbers of sequences given in brackets. Numbers at tree branches indicate the bootstrap values ( > 50%) from 1000 replicates. Bootstrap values over 99% are labeled with *, and those that were less than 50% are not shown. The scale bar represents the number of amino acid substitutions per residue.
-
Eighty-five clones, ranging from 790 to 884 bp, were obtained and identified as RNR homologs. Similarity analysis revealed that these sequences shared 30.7%– 99.6% identity at the nucleotide level and 47%–99% identity at the amino acid level. Their protein sequences had 67%–86% similarity with the closest relatives found in the GenBank database. Seventy-four nonredundant RNR amino acid sequences were selected for construction of a phylogenetic tree with known sequences of cyanophage isolates and host species. As shown in Figure 4A, five clusters were identified in the tree. East Lake sequences were either grouped within the largest S-CBS4-like cluster or the T7-like cluster. Most of East Lake sequences were closely related to Synechococcus viruses S-CBS4, S-CBP3, or S-CBP4. All of the three cultivated viruses were isolated from Chesapeake Bay(Huang et al., 2012). At the subcluster level, East Lake sequences could be further divided into EL 1–3 subgroups. RNR sequences from cyanophage isolates of different families formed independent clusters, such as the robust T7-like, T4-like, and S-CBS2/PSS2-like clusters. The T4-like cluster included two distinct subclusters, i.e., MM and FM, which were respectively formed with marine water and freshwater myoviruses.
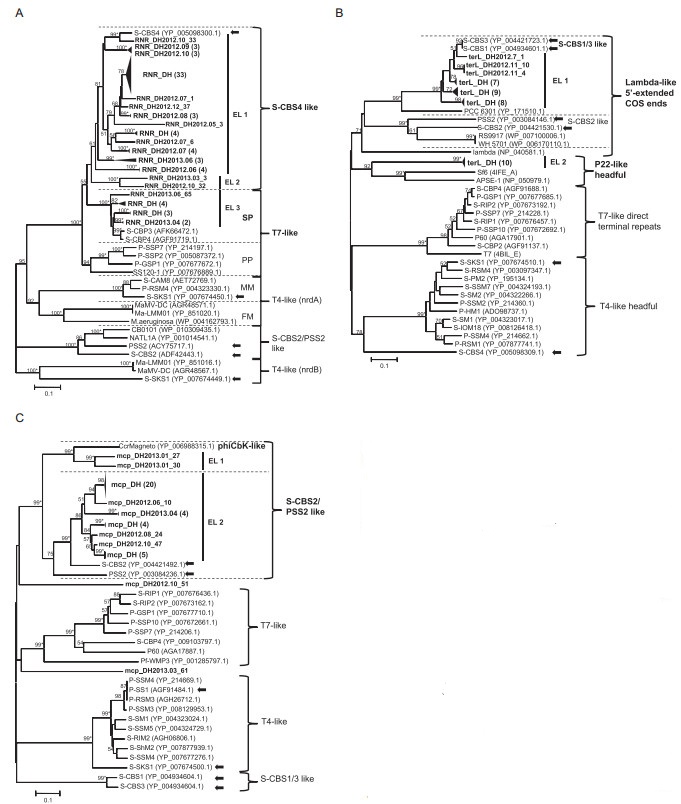
Figure 4. Neighbor-joining phylogenetic analysis of cyanosiphovirus-related sequences from East Lake. (A) Partial amino acid sequences of 74 RNR-related gen es from East Lake were aligned with those of isolated cyanophages and cyanobacteria and used for constructing a phylogenetic tree. SP: sequences related to Synechococcus viruses; PP: sequences related to Prochloroccus viruses; MM: sequences from marine cyanomyoviruses; FM: sequences from freshwater cyanomyoviruses. (B) Partial amino acid sequences of 37 terL-related genes from East Lake were aligned with th ose of phage isolates, cyanobacteria, and organisms with unknown structures and were used for construction of a phylogenetic tree. The end structure of virion DNA of known viruses is shown on the right of the tree. (C) Partial amino acid sequences of 38 mcp-related gen es from East Lake, aligned with those of phage isolates and used for construction of a phylogenetic tree. EL: gr oups containing only East Lake sequences (bold) with a bootstrap value over 60%. Black triangles indicate groups of East Lake sequences with high similarities, with numbers of sequences given in brackets. Arrows indicated cyanosiphovirus isolates. Numbers at tree branches indicate the bootstrap values (> 50%) from 1000 replicates. Bootstrap values over 99% are labeled with *, and those with values of less than 50% are not shown. The scale bar represents the number of amino acid substitutions per residue.
Fifty clones, ranging from 838 to 865 bp in length, were found to be homologous to terL genes. These sequences shared similarities between 38.3% and 100% at the nucleotide level and 9.5% to 100% at the amino acid level. Their protein sequences had amino acid identities ranging from 36% to 90% with the closest siphoviruses. Among them, 36 were closely related to the cyanosiphovirus subtype S-CBS1/S-CBS3, with identities between 83% and 90%; the other 14 were similar to bacteria siphoviruses, with identities between 36% and 42%. Next, we compared the 37 terL protein sequences from East Lake with those of phages for which end structures of the virion chromosome were determined or unknown(Figure 4B). These terL sequences formed four clusters according to the viral DNA end structure. East Lake sequences fell in two distinct clusters: the Lambda-like 5′-extended COS ends cluster(EL 1, 27 sequences)with four known cyanosiphoviruses, and the P22-like headful phage cluster(EL 2, 10 sequences).
We identified 38 clones from East Lake as mcp homologs of cyanosiphovirus subtype S-CBS2/P-SS2. The sizes of these mcp homologs varied from 597 to 739 bp and shared similarities from 22.5% to 99.9% at the nucleotide level and 13.4% to 100% at the amino acid level. Their protein sequences had 40%–72% similarity with their closest relatives. About 89% of sequences(34 out of 38)were closely related to the Synechococcus phage S-CBS2. The phylogenetic tree was constructed based on the alignments of 38 mcp protein sequences from this study and related sequences recovered from environmental and cultured phages(Figure 4C). Four robust clusters, i.e., S-CBS2/PSS2-like, S-CBS1/3-like, T4-like, and T7-like, were identified. The largest cluster, S-CBS2/PSS2, could be divided into two subclusters. Most of the East Lake sequences(34 out of 38, EL 2)were clustered together with Synechococcus phage S-CBS2 and Prochloroccus phage PSS2. For the remaining four sequences, two(mcp_DH2013.01_27 and mcp_DH2013.01_30)were within the phiCbK-like subcluster, and two(mcp_ DH2012.10_51 and mcp_DH2013.03_61)formed monophyletic branches and were distantly related to other sequences in the phylogenetic tree.
-
With the primer set of polB, 49 sequences were amplified and identified as phycodnavirus polB homologs, with lengths ranging from 428 to 666 bp. These sequences shared 28.1%–100% identity at the nucleotide level and 47.5%–100% identity at the amino acid level. Their protein sequences had amino acid identities ranging from 56% to 99% with the closest relatives. We used 43 nonredundant protein sequences from this study to construct a phylogenetic tree with close relatives(Figure 5A). In the tree, seven clusters were identified and covered all six known phycodnavirus genera: Prasinovirus, Raphidovirus, Coccolithovirus, Chlorovirus, Prymnesiovirus, and Phaeovirus. Most of East Lake clones(37/43)fell into the Prasinovirus cluster. Within this large cluster, three known subclusters, Fre Ⅰ(seven East Lake sequences), Fre-Mar(four East Lake sequences), and Fre Ⅱ(11 East Lake sequences)(Short and Short, 2008) and one novel subcluster(Fre Ⅲ, 14 East Lake sequences)were further resolved in this study.
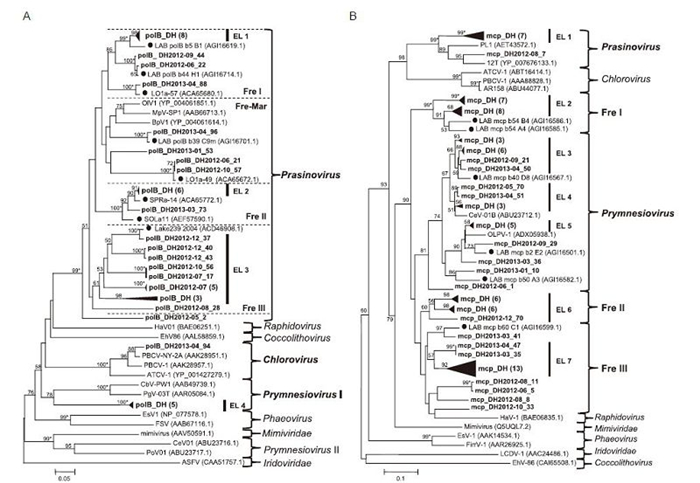
Figure 5. Phylogenetic analysis of phycodnavirus-related sequences from East Lake. (A) Partial amino acid sequences of 43 polB homologs from this study aligned with those of isolated phycodnaviruses and clones detected from other environments. (B) Partial amino acid sequences of 80 mcp homologs from this study aligned with those of isolated phycodnaviruses and clones detected from other environments. Genus names of known viral hosts are shown to the right of trees. Fre: cluster containing sequences from freshwater environments; Fre-Mar: cluster containing sequences from both freshwater and marine environments; EL: groups containing only East Lake sequences (bold) with the bootstrap value over 60%. Filled circles indicate published environmental sequences from freshwater systems. Bootstrap values (1000 replicates) over 50% are indicated at the nodes, and those over 99% are indicated by asterisks (*). The scale bar value shows the proportion of expected changes per site.
With the primer set used for mcp, 111 sequences were obtained from 12 samples homologous to phycodnavirus mcp genes, whose lengths varied from 374 to 582 bp. Similarity analysis revealed that these sequences shared 30.5%–100% nucleotide identity and 33.8%–100% amino acid identity. Translated protein sequences had amino acid similarities ranging from 43% to 89% with phycodnaviruses or clones in the GenBank nr database. The phylogenetic tree was constructed with 80 different protein sequences from East Lake, related sequences from other environments and cultured viruses with known hosts(Figure 5B). At least 10 clusters were identified by phylogenetic analysis, including all of the six known phycodnavirus genera as described above. Sequences from East Lake were clustered into five clusters, including Prasinovirus, Prymnesiovirus, Fre Ⅰ, Fre Ⅱ, and Fre Ⅲ. The Prasinovirus and Prymnesiovirus clusters were well-supported clusters that contained eight and 25 East Lake sequences, respectively. The Fre Ⅰ and Fre Ⅱ clusters, containing 15 and 13 East Lake sequences, respectively, were two stable environmental sequence-only clusters(bootstrap value > 60). In contrast, the Fre Ⅲ cluster was a weakly supported clade and contained 20 East Lake sequences and one clone from French lake(Zhong and Jacquet, 2014). However, we did not obtain any East Lake sequences grouped into Chlorovirus, Phaeovirus, Coccolithovirus, or Raphidovirus clusters.
Prevalence of phytoplankton viruses in East Lake
Diversity of cyanomyoviruses as determined by the g20 gene
Diversity of cyanopodoviruses as determined by the polAL gene
Diversity of cyanosiphoviruses as determined by the RNR, terL, and mcp genes
Diversity of phycodnaviruses determined by the polB and mcp genes
-
Through molecular techniques, our study revealed a high prevalence of phytoplankton viruses in East Lake.Sequences obtained from East Lake covered both cyanophages and phycodnaviruses. Cyanophages showed higher genetic diversity in summer and autumn than in winter, whereas no fluctuations in the diversity of phycodnaviruses were observed. These distinct temporal patterns of viral distribution indicated that cyanophages and their hosts may represent domain microbial communities in the East Lake ecosystem. The seasonal patterns of cyanophage communities were correlated with warmer temperatures at sampling times, during which phytoplankton were blooming(Xia et al., 2013). As a result, we concluded that water surface temperature was an important driver affecting the distribution patterns of cyanophage communities, rather than phycodnavirus communities, in East Lake. Although the exact roles that viruses play in host succession are unresolved, viral infection of competitively dominant phytoplankton species could lead to the so-called "killing the winner" phenomenon, which ultimately alters the community structure with other species(Thingstad, 2000; Winter et al., 2010).
To examine the diversity of phytoplankton viruses in East Lake, the obtained viral homologs were compared with sequences from both environmental and cultivated viruses. During the last decade, the diversities of cyanophages and phycodnaviruses have been examined in different marine and freshwater ecosystems(Parvathi et al., 2012; Clasen et al., 2013; Zhong and Jacquet, 2013). In this study, more than half(52%)of the sequences shared less than 80% amino acid similarity with published sequences in GenBank, indicating that the majority of phytoplankton viruses were quite distinct in this specific ecosystem in comparison to other environments. Interestingly, over 50% of cyanopodoviruses and over 90% of cyanosiphovirus homologous sequences from East Lake were closely related to those of cultured viruses from Chesapeake Bay(Sullivan et al., 2009; Huang et al., 2012), suggesting that these two aquatic ecosystems may have some common virus-host systems. This phenomenon could also be explained by potential bias created by the PCR primers, which were primarily based on sequences of cyanopodoviruses and cyanosiphoviruses from Chesapeake Bay. Nevertheless, over 86% of cyanomyovirus and phycodnavirus homologs were closely related to environmental sequences for which host are unknown. Thus, these data suggested that the majority of cyanomyoviruses and phycodnaviruses have not yet been discovered and could be major factors associated with phytoplankton mortality in East Lake(Zhong et al., 2013). Regardless of the diversity of East Lake sequences, most sequences were closely related to those from freshwater environments and formed distinct groups in the phylogenetic trees. However, there were some exceptions observed in this study; for example, some homologs were grouped closely with marine isolates, indicating the existence of common ancestors between these distinct environments.
Compared with g20, polAL, mcp, and polB genes, which are conserved in cyanomyoviruses, cyanopodoviruses, and phycodnaviruses, there are no such conserved genes for cyanosiphoviruses(Huang et al., 2012). Indeed, the genetic diversity of cyanosiphoviruses has not been explored sufficiently. By Clustal X analysis of sequences from our previous metagenome data and the five known cyanosiphovirus isolates, three degenerate primer sets targeting the RNR gene of the S-CBS4 subtype, terL gene of the S-CBS1/S-CBS3 subtype, and mcp gene of the S-CBS2/PSS2 subtype were designed in this study. With these three primer sets, S-CBS4 subtype viruses were successfully detected in 10 of 12 months, S-CBS1/ S-CBS3 subtype viruses were successfully detected in 8 months, and S-CBS2/PSS2 subtype viruses were successfully detected in 10 months. BLAST and phylogenetic analysis of these sequences revealed that most of these sequences were closely related to the known cyanosiphoviruses of three subtypes. Therefore, the three novel primer sets in this study could be used as molecule markers to study the genetic diversity and prevalence of cyanosiphovirus communities in freshwater systems.
Previous studies have suggested that the genetic diversity of phytoplankton viruses depends on the diverse interactions between viruses and hosts(Larsen et al., 2008; Wang et al., 2010). In this study, a large proportion of cyanomyovirus-and phycodnavirus-related sequences were distantly related to known viruses and fell within the environmental-only clusters in phylogenetic trees. These results supported the existence of differing host-virus interaction systems in freshwater compared with their marine counterparts. Considering the relatively few phytoplankton viruses that have been isolated, especially from freshwater systems, it is difficult to predict the hosts of these viral sequences. However, our data provided information for predicting plausible hosts for some viruses. For example, the majority of sequences related to cyanopodoviruses and cyanosiphoviruses were most similar to marine viruses infecting Synechococcus. Therefore, it is reasonable to presume that Synechococcus-like cyanobacteria may be important hosts for many cyanophages in East Lake. In addition, the clone polB_ DH2013-04_94 had 99% amino acid similarity with Paramecium bursaria Chlorella virus 1(PBCV-1)(Zhang et al., 2001), indicating that this sequence-related virus may also infect Paramecium bursaria Chlorella. These observations may be helpful for future virus isolation.
In summary, to the best of our knowledge, this is the first year-long investigation determining the prevalence and diversity of the vast majority phytoplankton viruses in a freshwater environment, simultaneously including both cyanophages of three families and viruses of Phycodnaviridae. Our results provided a broad analysis of viral diversity in East Lake and will be helpful for future control of harmful phytoplankton blooms. Nevertheless, it is important to note these primer sets cannot recover all phytoplankton viruses. There may be an underestimation of phytoplankton virus communities in East Lake. Thus, other methods should be used in combination for exploration of phytoplankton viruses in future studies.
-
We acknowledge financial support from Knowledge Innovation Program of the Chinese Academy of Sciences(KSCX2-YW-Z-0954, KSCX2-EW-Z-3).
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.
-
ZL Shi and MN Wang designed this experiment and analyzed the date. XY Ge, YQ Wu, XL Yang, YJ Zhang and B Tan helped to collect samples. MN Wang wrote this paper. All authors read and approved the final manuscript.

 DownLoad:
DownLoad: