










HTML
-
Innate immune signaling pathways are activated when pathogen-associated molecular patterns (PAMPs) are recognized by host pattern-recognition receptors (PRRs) (Brubaker et al., 2015). A major consequence is the activation of multiple transcription factors, including nuclear factor-kappa B (NF-κB) and interferon regulatory factors (IRFs), which drive the expression and secretion of inflammatory cytokines and type Ⅰ IFNs that restrict the proliferation and dissemination of infecting microbes. Type I IFN-mediated signaling triggers phosphorylation of signal transducers and activators of transcription 1 (STAT1) and STAT2. STAT1, STAT2 and IRF9 form a complex termed Interferon Stimulation Gene Factor 3 (ISGF3), which is responsible for the induction of hundreds of IFN-Stimulated Genes (ISGs) (Platanias, 2005). Specifically, tyrosine701 (Y701) of STAT1 is phosphorylated upon IFN stimulation, and is essential for the induction of a broad range of ISGs (Platanias, 2005).
The canonical IKKs, including IKKα and IKKβ, form a complex with the adaptor protein NEMO that is required for NF-κB activation. IKKα and IKKβ contain an N-terminal kinase domain, a central leucine-zipper and helix-loop-helix domain, and C-terminal NEMO-binding domain (NBD). The non-canonical IKKs, i.e., TANK binding kinase 1 (TBK1) and IKKε (also known as IKKi) (Shimada et al., 1999), share a domain structure similar to IKKα and IKKβ. However, TBK1 and IKKε do not have NBD, and presumably do not associate with NEMO (Verhelst et al., 2013). This review will focus on IKKε, while the closely related TBK1 kinase will be referred to whenever relevant function is considered.
-
IKKεis required for antiviral immune response IKKε and TBK1 appear to play similar function in phosphorylating IRF3 and IκBα (Peters et al., 2000; Fitzgerald et al., 2003; Sharma et al., 2003), which is consistent with the finding that IKKε and TBK1 share similar substrate phosphorylation motif (Hutti et al., 2012). Nonetheless, there are some differences between IKKε and TBK1. Although initial studies demonstrated that TBK1 and IKKε can phosphorylate IRF3, work using gene knockout cells and mice show that TBK1, but not IKKε, is essential for IFN induction in response to viral infection (Mcwhirter et al., 2004). In contrast to TBK1 that is constitutively expressed in most cell types, the expression of IKKε is limited to specific cell types (e.g., lymphocytes) and is induced by PMA, LPS and many inflammatory cytokines (Shimada et al., 1999; Peters et al., 2000). Knockout of TBK1 in mice leads to embryonic lethality (Bonnard et al., 2000; Hemmi et al., 2004), whereas IKKε-deficient mice are viable and born with Mendelian heredity (Hemmi et al., 2004; Tenoever et al., 2007). These results agree with the conclusion that IKKε and TBK1 control non-overlapped signaling events. For example, IKKε, but not TBK1, is activated by type Ⅰ IFNs to phosphorylate STAT1 and induce the expression of a subset of ISGs (Tenoever et al., 2007; Perwitasari et al., 2011). Considering that IKKε and TBK1 have identical substrates in vitro (Hutti et al., 2012), the different substrate specificity of IKKε and TBK1 in vivo likely stems from other regulatory factors. One intriguing example is TRIM6 that catalyzes the assembly of K48-linked polyubiquitin chain to specifically activate IKKε upon type Ⅰ IFN-stimulation (Rajsbaum et al., 2014).
Despite that overexpressed IKKε was initially reported to phosphorylate IκBα (Shimada et al., 1999; Peters et al., 2000), subsequent studies revealed that loss of IKKε did not affect IκBα degradation. Instead, TBK1 and IKKε are able to phosphorylate the transcription factors IRF3 and IRF7 that is required for type Ⅰ IFN production (Sharma et al., 2003; Hemmi et al., 2004). Moreover, IFN induction in IKKε-deficient mice and primary cells was not reduced upon influenza virus challenge (Tenoever et al., 2007). Taken together, it seems that IKKε is dispensable for influenza virus-induced IFN production. Alternatively, IKKε may play a redundant role with other molecules, i.e. TBK1, in IFN production during influenza virus infection. The role of IKKε in IFN production induced by other types of viruses awaits further investigation. On the other hand, IKKε is required for the efficient induction of a subset of ISGs upon type Ⅰ IFN stimulation.
-
Compared to wild-type mice, IKKε knockout mice were more susceptible to influenza virus infection (Tenoever et al., 2007). Surprisingly, IKKε knockout mice and primary cells produced IFNs comparable to wild-type counterparts upon influenza virus challenge (Tenoever et al., 2007). It was found that loss of IKKε reduced the induction of a subset of ISGs, including ISG54 (or IFIT2), ADAR1, IFIT3 and Mx1. Interestingly, some ISGs, such as IRF7 and ISG15, were expressed in an IKKε-independent manner in response to influenza virus infection. Thus, IKKε is required for the efficient induction of ISGs upon IFN stimulation. Similarly, IKKε has been shown to be required for West Nile Virus (WNV)-induced IFIT2 production (Perwitasari et al., 2011). Using STAT1 S708 phospho-specific antibody, Perwitasari et al. unambiguously demonstrated that IKKε was necessary for STAT1 S708 phosphorylation upon IFN treatment. In contrast, the phosphorylation of Y701 and S727 was not dependent on IKKε. During WNV infection, IRF3 activation clearly preceded STAT1 S708 phosphorylation, consistent with the notion that IKKε is activated by IFN stimulation. Conversely, IFN signaling is necessary for STAT1 phosphorylation by IKKε in response to WNV infection, because the phosphorylation of STAT1 at S708 was abolished in cells deficient in IRF3 and IFN receptor. Moreover, IFIT2 induction was significantly reduced in IKKε knockout mice such that IKKε knockout mice were more susceptible to WNV infection than wild-type mice (Perwitasari et al., 2011). Recently, IKKε was found to be involved in IFN-induced Usp25 expression and IRF7 was required for the induction of Usp25 (Lin et al., 2015; Ren et al., 2016). IKKε and TBK1 played redundant roles in this process, since either the expression of IKKε or TBK1 could support the induction of Usp25 upon IFN treatment. Whether the IFN-IRF7 signaling axis is involved in the induction of other ISGs awaits further study. Another interesting question remained to be addressed is whether TBK1 is also required for the efficient induction of other ISGs upon IFN treatment. Nonetheless, these findings collectively indicate that IKKε is critical to elicit a full breadth of IFN-mediated antiviral response via inducing ISG expression.
As summarized, substantial evidences indicate that the phosphorylation of STAT1 at S708 by IKKε is required for the induction of IKKε-dependent ISGs upon IFN stimulation. This conclusion is further explored and validated by several studies (Perwitasari et al., 2011; Rajsbaum et al., 2014). For example, TRIM6, which is necessary for type Ⅰ IFN-induced IKKε activation, is also critical for the induction of IKKε-dependent ISGs. This observation brings an intriguing question: Why IKKε only activates a subset of ISGs? Computational analysis revealed that IKKε-independent ISGs contain a purine-rich sequence in IFN-stimulated response elements (ISREs), which is absent in IKKε-dependent ISGs (Tenoever et al., 2007). The purine-rich motif may enhance the binding of STAT2 and the transcription of IKKε-independent ISGs. Based on this observation, it was postulated that, in the absence of the purine-rich sequence in IKKε-dependent ISGs, a more stable interaction between STAT1-STAT2-IRF9 (ISGF3) will be required for the binding of ISGF3 to ISRE-containing promoters to initiate an effective transcription. Thus, the phosphorylation of STAT1 by IKKε at S708 facilitates the formation of a more stable ISGF3 to enable the transcription of IKKε-dependent ISGs. This hypothesis remains to be experimentally tested.
Upon type Ⅰ IFN treatment, the phosphorylation of STAT1 S708 was detected at time points later than that of STAT1 Y701 (Perwitasari et al., 2011). Furthermore, the phosphorylation of S708 occurred only after Y701 was dephosphorylated, suggesting that the phosphorylation of these two sites is mutually exclusive (Perwitasari et al., 2011). It was previously reported that de novo protein synthesis is required for the phosphorylation of STAT1 S708 induced by type Ⅰ IFNs (Perwitasari et al., 2011). One possibility is that the newly synthesized ISG(s) is necessary for STAT1 S708 phosphorylation by IKKε. Since TRIM6 was not induced by IFNs, the IFN-induced factors that are required for IKKε to phosphorylate STAT1 S708 remain to be identified. On the other hand, how phosphorylation of STAT1 S708 affects the ISRE-binding activity of ISGF3 calls for further investigation. The detailed structural analysis of STAT1 carrying phosphorylated S708 will undoubtedly provide critical insights into the interaction between a transcription factor with its cognate responsive promoters. Another related question is whether the difference in the expression of IKKε-dependent and -independent ISGs stems solely from the action of the S708 phosphorylated STAT1, i.e. whether alternative substrates or signaling events downstream of IKKε also contribute to the expression of those IKKε-dependent ISGs. The alternative substrates or signaling events are poorly defined (Figure. 1).
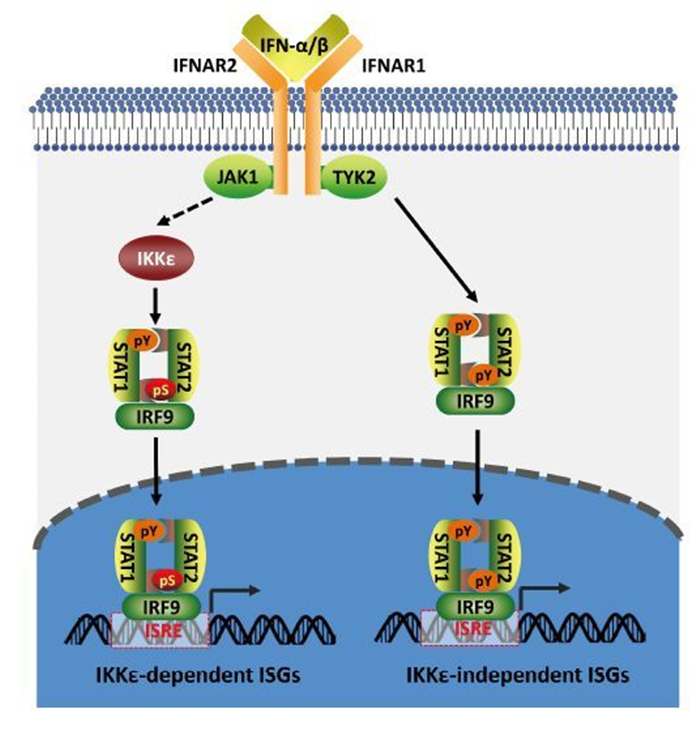
Figure 1. IKKε is required for the efficient induction of ISGs upon IFN stimulation. Type I interferons (IFNs), including IFN-α and IFN-β bind type Ⅰ IFN receptor (IFNAR) on the surface of human cells. IFNAR consists of two subunits, IFNAR1 and IFNAR2, which are associated with tyrosine kinase 2 (TYK2) and JAK1, respectively. Activation of JAK1 and TYK2 upon IFN stimulation leads to tyrosine phosphorylation of STAT1 and STAT2. Together with IRF9, they form the STAT1-STAT2-IRF9 complex termed ISGF3. The ISGF3 complex translocates into the nucleus and binds IFN-stimulated response elements (ISREs) to initiate gene transcription. IFN treatment activates IKKε that phosphorylates STAT1 at S708. The phosphorylation of STAT1 by IKKε promotes the transcription of a subset of ISGs (IKKε-dependent ISGs).
-
In contrast to the antiviral activity of IKKε against RNA viruses (e.g., influenza virus), loss of IKKε in mice dramatically reduced the persistent infection of a model DNA virus (Zhang et al., 2016), murine gamma herpesvirus 68 (γHV68). IKKε knockout mice produced significantly higher levels of antiviral CD8 T cell response upon γHV68 challenge. This observation is consistent with the notion that T cell immunity is the major player to contain viral persistent infection, in the form of latent infection for herpesviruses. Mechanistically, IKKε was identified as a kinase of nuclear factors of activated T cells (NFATs) and phosphorylated NFAT proteins to restrict the activation of NFAT transcription factors (Figure 2). Key transcriptional factors of T cell activation (Muller and Rao, 2010), NFAT proteins contain an amino-terminal transactivation domain, a regulatory domain, a DNA-binding domain and a carboxyl-terminal domain that often harbors an additional transactivation domain (Muller and Rao, 2010). The regulatory domain can be phosphorylated by various kinases, including casein kinase 1, glycogen synthase kinase 3β, and the dual-specificity tyrosine phosphorylation regulated kinase (DYRK) (Muller and Rao, 2010). In resting cells, the phosphorylation of NFAT by these kinases excludes NFAT from the nucleus. Upon T cell activation, calcium influx activates numerous calcium-dependent enzymes. The most extensively studied calcium-dependent enzyme, the calcineurin phosphatase dephosphorylates NFAT, resulting in NFAT nuclear translocation and up-regulated expression of NFAT-responsive genes. Roles of most NFAT kinases, except DYRK1A, in suppressing NFAT signaling activity in vivo has not been reported. Interestingly, the transgenic expression of DYRK1A in mice was sufficient to decrease the protein level of DSCR1 (also known as RCAN1), an NFAT-dependent gene, and induce vascular defects (Arron et al., 2006). The deficit of vascular system agrees with that NFAT activation is crucial for endothelial cell growth and proliferation (Johnson et al., 2003). These results indicate that DYRK1A suppresses NFAT activity in vivo. Conversely, loss of IKKε releases its restriction of NFAT signaling and enhances T cell activation, supporting its function as an NFAT kinase. T cell activation by receptor ligation or pharmacological activation (e.g., PMA) induced the activation of IKKε (Yu et al., 2015; Zhang et al., 2016). Activated IKKε in T cells phosphorylated NFAT, but not S708 of STAT1 (Zhang et al., 2016), suggesting that IKKε relays signal transduction to mount distinct transcriptional responses in a manner dependent on cell type or apical signal. Moreover, viral latent infection and tumor cell challenge induced the activation of IKKε, thus suppressing T cell immunity against viral or tumor antigens (Zhang et al., 2016). These results imply the potential to boost the antiviral and antitumor activity of CD8 T cells by inhibiting IKKε, analogous to the PD-1/PD-L1 blockage immunotherapy (Zitvogel and Kroemer, 2012). Collectively, these findings support the conclusion that IKKε serves as a negative feedback kinase to restrict NFAT activation and T cell immune response, suggesting that IKKε can be a potential therapeutic target to improve antiviral and antitumor immunotherapy.
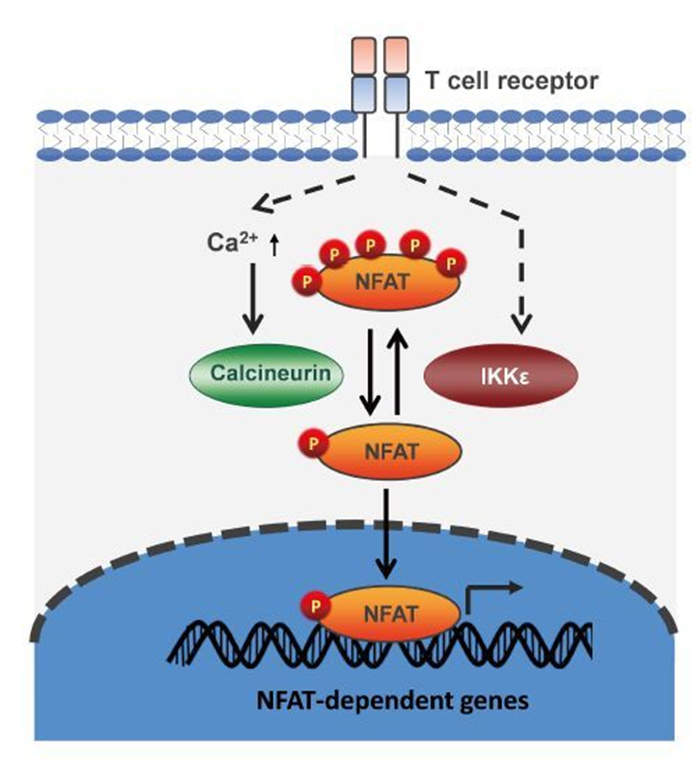
Figure 2. IKKε phosphorylates NFAT and restricts NFAT activation during T cell activation. In resting cells, NFAT is hyperphosphorylated and resides in the cytoplasm. Upon T cell activation, increased calcium leads to the activation of the calcineurin phosphotase, which dephosphorylates NFAT, promotes NFAT nuclear translocation and activates gene expression. T cell activation stimulates IKKε, which phosphorylates NFAT and counteracts on NFAT signaling activation. Thus, IKKε restricts T cell activation as a negative feedback regulator.
The dramatically reduced γHV68 latent infection in IKKε-deficient mice is reminiscent of the observation that blockade of type Ⅰ IFNs reduced LCMV latent infection in mice (Teijaro et al., 2013; Wilson et al., 2013). Chronic IFN signaling has been associated with disease progression in LCMV persistent infection. Treatment with antibody blocking IFNAR1 reversed these pathological phenotypes and diminished LCMV persistent infection. The reduced LCMV persistent infection depended on CD4 T cells and IFNγ, while the antibody response and CD8 antiviral cytotoxicity were not affected by IFN blockade (Teijaro et al., 2013; Wilson et al., 2013). Because type Ⅰ IFNs activate IKKε and IFNγ is a known NFAT-dependent gene (Kiani et al., 2001), it is tempting to speculate that the elevated antiviral T cell immunity of IFN blockade is partly achieved via ablating the IKKε-mediated inhibition of NFAT signaling. Such a molecular link may provide a mechanistic explanation underpinning the effect of IFN blockade on LCMV infection and pathogenesis.
While IKKεdeficiency elevates CD8 T cell immune response in vivo, loss of TBK1 in mice elevated CD4 T cell Immune response. T cell-specific ablation of TBK1 promoted CD4 T cell activation, and caused retention of effector T cells in the draining lymph node in an experimental autoimmune encephalomyelitis (EAE) mouse model (Yu et al., 2015). At the molecular level, TBK1 stimulated AKT ubiquitination and degradation, thereby down-regulating AKT signaling in CD4 T cells. Thus, the ablation of TBK1 increased CD4 T cell activation and reduced effector T cell exit from the draining lymph nodes. Treatment of EAE mice with Amlexanox (a TBK1 inhibitor) alleviated these symptoms, suggesting that TBK1 is a potential therapeutic target for neuroinflammation and related diseases (Yu et al., 2015). The function of IKKε in CD4 T cell Immune response remains undetermined, but it is tempting to speculate that IKKε deficiency may enhance CD4 T cell activation. However, this needs to be examined with appropriate in vivo mouse studies.
-
It was previously shown that type Ⅰ IFNs activate IKKε (Tenoever et al., 2007). A subsequent study showed that treatment with type Ⅱ and Ⅲ IFNs also activated IKKε (Perwitasari et al., 2011). Despite these interesting observations, the detailed mechanism by which IKKε is activated by IFNs remains unknown. Recently, an interesting study by Rajsbaum et al.has made a significant step towards understanding how IKKε is activated by type Ⅰ IFNs (Rajsbaum et al., 2014). It was found that TRIM6 interacted with IKKε and was required for the expression of IKKε-dependent ISGs induced by IFNs. TRIM6, in the presence of the E2 ubiquitin-conjugating enzyme UbE2K, catalyzed the synthesis of unanchored K48-linked polyubiquitin chains that in turn triggered IKKε activation. This study, for the first time, provides a mechanistic insight into the activation of IKKε. One surprising observation is that the free K48-linked ubiquitin chain serves as a second messenger to activate IKKε. Previously, it has been shown that unanchored K63-linked ubiquitin chain is required for the activation of theintracellularRNA sensor RIG-I (Zeng et al., 2010). These studies collectively support the conclusion that the free polyubiquitin chain is a second messenger to facilitate antiviral immune response. Specific to IKKε activation by TRIM6, the authors also observed a very interesting cellular structure, which was dubbed "Ub factories". Within these "Ub factories", TRIM6 recruits IKKε and Ub to facilitate the synthesis of free polyubiquitin chains that instigate IKKε activation. The question to follow was how TRIM6 is activated. The authors speculated that JAK1 may phosphorylate TRIM6, which leads to its activation. And this hypothesis remains to be experimentally examined.
In addition to IFN treatment, T cell receptor engagement and calcium influx potently activated IKKε in T cells (Peters et al., 2000; Yu et al., 2015; Zhang et al., 2016). Interestingly, IKKε activated by calcium mobilization in T cells is distinct from that activated by IFNs. During T cell activation, activated IKKε potently phosphorylated NFAT transcription factors and inhibited T cell immune responses, while STAT1 S708 phosphorylation was not detected. It remains to be determined whether IKKε can phosphorylate NFAT upon IFN treatment and whether TRIM6 and free K48-linked polyubiquitin chains are also critical for IKKε activation in T cells. Nevertheless, these findings suggest that the substrate specificity of activated IKKε is context-dependent, which may include cell type and the nature of apical activating signal. For example, IKKε may be activated by distinct mechanism upon IFN treatment or T cell activation by TCR ligation or pharmacological activation. Whether distinct forms of activated IKKε exist or tissue-specific cofactors enable substrate specificity of IKKε remains to be determined. Investigation into this intriguing question will shed light on the mode of activation of IKKε under diverse physiological conditions.
-
IKKε and TBK1 are critical for host antiviral immune responses. As obligate intracellular pathogens, viruses have evolved elaborate strategies to evade host immune defense by targeting IKKε and TBK1. Viral immune evasion through targeting TBK1 has been reviewed extensively elsewhere (Zhao, 2013; Hasan and Yan, 2016). Ebola virus VP35 blocks virus-induced IRF-3 phosphorylation and inhibits the IRF3-dependent gene expression (Basler et al., 2003; Cardenas et al., 2006). Subsequently, it was found that VP35 interacted with both ΙΚΚε and TBK-1, and blocked their interactions with IRF3, thereby preventing IRF3 phosphorylation (Prins et al., 2009). Severe fever with thrombocytopenia syndrome (SFTS) is a febrile illness caused by the emerging phlebovirus. The SFTS virus (SFTSV) nonstructural protein formed inclusion bodies in infected cells and sequestered IKKε and IRF3 into inclusion bodies to eliminate their antiviral activity (Wu et al., 2013). Several Paramyxoviruses V proteins interact with TBK1 and IKKε to inhibit IRF3 activation. Intriguingly, V proteins encoded by mumps virus (MuV), human parainfluenza virus 2 (hPIV2), and parainfluenza virus 5 (PIV5) were phosphorylated by TBK-1/ IKKε, reducing the expression of V proteins. Thus, V proteins mimic IRF3 in both its phosphorylation by TBK1/IKKε and the subsequent degradation (Lu et al., 2008), serving as a decoy substrate to block IRF3 phosphorylation and activation. ORF45 of Kaposi's sarcoma-associated herpesvirus (KSHV) employs a similar substrate mimicry strategy to antagonize IRF3 phosphorylation and IFN induction. Consistent with the decoy substrate idea, ORF45 was phosphorylated more efficiently than IRF7 by IKKε and TBK1. Thus, KSHV ORF45 serves as an alternative but more efficient substrate to suppress IRF7 phosphorylation and activation (Liang et al., 2012). Additionally, a KSHV-encoded microRNA, miR-K12-11, down-regulated IFN signaling through targeting IKKɛ. Expression of miR-K12-11 attenuated IFN signaling by decreasing IKKɛ-mediated IRF3/IRF7 phosphorylation and by inhibiting the expression of IKKɛ-dependent ISGs (Liang et al., 2011).Hepatitis C virus NS2 protease associated with IKKε and TBK1 and interrupted the kinase activity to block IFN production (Kaukinen et al., 2012). Recently, Nipah virus matrix (NiV-M) protein was found to interact with TRIM6 and promote TRIM6 degradation (Bharaj et al., 2016). TRIM6 synthesizes free K48-linked polyubiquitin chain that is required for IKKε activation and the induction of IKKε-dependent ISGs. Indeed, NiV-M expression reduced levels of unanchored K48-linked polyubiquitin chains associated with IKKε, thereby crippling IKKε activation and subsequent IFN-mediated responses. The matrix proteins of Ghana, Hendra and Cedar viruses have evolved similar strategies to antagonize IFN responses (Bharaj et al., 2016). These studies not only elucidate viral strategies that evade host immune defense mechanisms of IFN signaling, but also provide molecular insight into the regulation by which host IFN signaling can be dynamically regulated in viral infection and host immune defense.
-
IKKε was initially identified as a breast cancer oncogene in a shRNA library screen. Subsequently, accumulating studies implicated IKKε in the development of diverse human cancers of the prostate, liver, ovarian and skin (Boehm et al., 2007; Guo et al., 2009; Wang et al., 2013; Challa et al., 2016; Peant et al., 2016). Most notably, IKKε promoted oncogenesis through its intrinsic activity in activating NF-κB and AKT (Xie et al., 2011; Wang et al., 2013). Roles of IKKε in tumor development have been extensively reviewed elsewhere (Clement et al., 2008; Shen and Hahn, 2010; Verhelst et al., 2013) and will not be discussed here. Rather, we will briefly discuss the emerging possibility to target IKKε for immunotherapy.
Recent studies suggest that strategies targeting IKKε for inhibition are likely viable to enhance the antiviral and antitumor immunity. IKKε is activated by proinflammary cytokines such as TNF-α, IL-1β and IL-6. Chronic inflammation underpins the development of a broad spectrum of human cancers, and the activation of IKKε by these inflammatory cytokines may suppress the antitumor immune responses. Additionally, IKKε expressed in tumor cells promotes survival and proliferation, facilitating multiple steps of the tumorigenic process. Thus, inhibiting IKKε will not only thwart tumorigenesis per se, but also enhance the antitumor immunity of T cells to restrict tumor development and metastasis.
Worldwide, seven human tumor viruses have been found to cause around 15% of human cancers (McLaughlin-Drubin and Munger, 2008; Moore and Chang, 2010). Persistent viral infection triggers IKKε activation that suppresses the antiviral and antitumor activity of CD8 T cells (Zhang et al., 2016). Theoretically, inhibitors of IKKε will boost NFAT activation and the killing activity of CD8 T cells. Similar strategies have been approved to be a great success by the application of immune checkpoint inhibitors (Postow et al., 2015).
Currently, BX795 and Amlexanox are widely used to inhibit IKKε activity in cultured cells (Clark et al., 2009; Reilly et al., 2013). Similar to other kinase inhibitors, these compounds have apparent off-target effect and often toxic. Moreover, Amlexanox suffers from low potency in vitro with an IC50 of 20-50 mmol/L, which makes it difficult to effectively inhibit IKKε in vivo. MRT67307 is a modified version of BX795 with improved specificity. MRT67307 can efficiently inhibit TBK1 and IKKε, but had no effect on JNK and p38 MAPK that were inhibited by BX795 (Clark et al., 2010). A series of azabenzimidazole derivatives were shown to potently inhibit TBK1 and IKKε (Wang et al., 2012). The in vivo effect of these compounds remains to be determined. The fact that TBK1 and IKKε demonstrate kinase activity toward an identical array of substrates in vitro (Hutti et al., 2009; Hutti et al., 2012) makes it a challenge to differentially target TBK-1 and IKKε in vivo. Considering the high identity of amino acid sequence shared by TBK-1 and IKKε, it is possible that IKKε adopts a structure similar to TBK-1 that has been solved by crystallography. Future detailed structural analysis of IKKε will pave the way for the development of IKKε-specific inhibitors.
-
The function of IKKε as an oncogene in cancer is more broadly appreciated, while its roles in IFN-mediated immune defense or T cell response are limited. This is largely due to the very few substrates identified to date. STAT1 phosphorylation by IKKε at S708 significantly advanced our understanding concerning the role of IKKε in IFN signaling, while NFAT phosphorylation by IKKε defined a surprising negative regulatory function of IKKε in T cell immune responses. Recently, it was shown that IKKε contributes to the maintenance of Th17 cell through phosphorylating GSK3α (Gulen et al., 2012). Specifically, IKKε was activated by IL-1 and phosphorylated S21 of GSK3α. As such, IKKε inactivated GSK3α to promote IL-1-induced AKT-mTOR activation, a signaling pathway critical for Th17 cell maintenance. This study implies that IKKε is a potential therapeutic target to treat IL-17-dependent autoimmune diseases.
IKKε substrate specificity was examined by in vitro kinase assay using a randomly synthesized peptide library. This work identified a phosphorylation motif (x-x-x-Y/F/P/M-x-pS-L/I/M/F-x-Y/W/F-x, with x being any amino acid) as IKKε substrate. Computational search guided by this deduced sequence of phosphorylation motif yielded a large number of potential substrates of IKKε (Hutti et al., 2009), a few of which were validated. Interestingly, IkBα, one of the best-defined IKKε substrates, doesn't contain the optimal motif. Additionally, phosphorylation sites of STAT1 and NFAT proteins do not conform to the consensus motif derived from the peptide library screen. Conceivably, the list of IKKε substrates will continue to expand with our increasing understanding in IKKε function. In doing so, it is plausible to systematically identify the substrate of IKKε by phosphoproteomics, which has been applied to dissect TBK1-mediated phosphorylation (Kim et al., 2013). It is anticipated that our knowledge of the key roles of IKKε in fundamental biological processes will explode along with the identification and functional characterization of substrate phosphorylation by IKKε.
-
IKKε is critically involved in diverse biological processes, including innate immune signaling, T cell activation, inflammation, tumorigenesis and metabolic diseases (Chiang et al., 2009). As summarized in this short review, significant progress has been made to understand roles of IKKε in signal transduction entailing innate and adaptive immunity. IKKε activation induced by IFN treatment leads to STAT1 phosphorylation and the induction of IKKε-dependent ISGs. During T cell activation, IKKε is stimulated to phosphorylate NFAT proteins and restrict T cell activation. To counteract the antiviral effect of IKKε, viruses have evolved various strategies to target IKKε and inhibit IKKε-mediated antiviral immunity. It is of great interest to develop IKKε modulators to treat chronic infectious diseases, autoimmune disorders and inflammation-related malignancies such as cancer. More studies are needed to elucidate the specific molecular mechanism governing IKKε activation under distinct physiological stimulations and pathological conditions.
-
This work was supported by the Joint Funds of the National Natural Science Foundation of China (U1603126) (to Z. X.).
-
The authors declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: