














-
Dear Editor,
Noroviruses are positive-sense, single-stranded RNA viruses belonging to Caliciviridae and account for more than 50% of all acute gastroenteritis (AGE) outbreaks worldwide and cause an estimated 200, 000 deaths per year among children < 5 years of age, primarily in developing countries (Hall et al.2012; Glass et al.2009).The norovirus genome contains three open reading frames (ORFs). ORF1 encodes nonstructural proteins, including an RNAdependent RNA polymerase (RdRp), while ORF2 and ORF3 encode the major (VP1) and minor (VP2) structural proteins, respectively.Based on the sequences of their VP1 genes, noroviruses are classified into at least seven genogroups (GI-GVII) and more than 30 genotypes (Vinjeé 2015).GI and GII viruses are responsible for most human infections.
Over the past 2 decades, GII.4 viruses have been the most common genotype to cause norovirus outbreaks worldwide, and new GII.4 variants have emerged approximately every 2-3 years through accumulations of mutations and intra-genotype recombination (Lindesmith et al. 2012;Eden et al.2013).During the 2014-2015 winter season, a new GII.17 variant emerged and became the predominant virus in China and parts of Asia, through changes in blockade antibody epitopes and the possible acquisition of a mutated polymerase (Chan et al.2015). However, during 2016-2017, a GII.P16-GII.2 strain reemerged as the major cause of norovirus outbreaks in China and other countries (Ao et al.2017; Bidalot et al.2017; Thongprachum et al.2017; Niendorf et al.2017; Cannon et al.2017), possibly driven by mutations in its nonstructural proteins, particularly the polymerases (Tohma et al. 2017;Ao et al.2018).This necessitates extensive epidemiological studies on GII.2 noroviruses causing both outbreaks and sporadic cases.
In this study, a total of 190 fecal samples associated with 37 norovirus outbreaks from December 2016 to July 2017 in the Fengtai District, Beijing City, were analyzed, using the process developed by the US CDC (Cannon et al. 2017).Thirty-six of 37 outbreaks were caused by the reemerging GII.P16-GII.2 norovirus, while three samples from one outbreak contained the GⅡ.P2-GII.2 norovirus. The analysis of the partially genotyped VP1 sequences (275 nt) of three GII.P2-GII.2 strains (BJFTJYX, BJFTZTX, and BJFTNJY) demonstrated 100% nt identity to the sequence of the reemerging GII.P16-GII.2 norovirus predominant in 2016-2017(Ao et al.2018), but the sequences were distinct from previous GII.P2-GII.2 strains, suggesting the presence of a new GII.P2-GII.2 strain.Therefore, we investigated the evolutionary and genetic characteristics of this strain.The capsid-encoding genes from three GII.P2-GII.2 strains were amplified from three samples using nested RT-PCR, yielding an approximately 2.5-kb amplicon, as described previously (Ao et al. 2018).The complete genome of one representative GII.P2- GII.2 strain BJFTJYX was amplified and sequenced.All sequences obtained in this study have been deposited in GenBank (Accession Numbers MH158635 and MH671553-MH671554).
The time-scale evolutionary trees of 148 GII.2 complete VP1 sequences and 79 partial GII.P2 RdRp sequences were estimated using the strict clock model, GTR+G/HKY+G substitution model, and Bayesian skyline coalescent model implemented in BEAST software (version 1.8.2).Markov chain Monte Carlo (MCMC) sample chains were run for 4×108 and 2.5×108 steps for the VP1 and RdRp genes, respectively, sampling both every 10, 000 generations.The VP1 gene phylogenetic tree could be grouped into three major clusters, according to their polymerase genotypes and sequence divergences, one each for the reemerging GII.P16-GII.2(2016-2017), GII.P16-GII.2(2008-2015), and GII.P2-GII.2(2004-2010) strains.BJFTJYX, BJFTZTX, and BJFTNJY formed peripheral branches in the cluster for the re-emerging GII.P16-GII.2 (2016-2017), which were closely related to the lineage of the 2011-2012 GII.P16-GII.2 strains, including the HS255/USA/2011(KJ407074.2) and Miyagi1/Japan/2012 (LC145787.1) strains.However, they were separated from the previous cluster of the GII.P2-GII.2 (2004-2010) strains (Fig. 1A).The divergence time analysis showed that the most recent common ancestor (MRCA) of the emerging GII.P2-GII.2 strain appeared in 2011-2012, whereas that of the GII.P16-GII.2 strains reemerging in 2016-2017 appeared in 2013(Fig. 1A), indicating that the appearance of the emerging GII.P2- GII.2 strain was earlier than that of the reemerging GII.P16-GII.2 viruses predominant in 2016-2017.These data suggest that this virus did not originate from the previous GII.P2-GII.2 strains but probably resulted from a novel recombination event.The RdRp gene phylogenetic tree could be divided into four lineages based on their evolutionary distances and detected time (Fig. 1B). Lineage 1 can be further split into two sub-lineages.The emerging GII.P2-GII.2 strain BJFTJYX belonged to lineage 1 and sub-lineage 2, which also included three GII.P2-GII.2 strains identified from July to October 2016 in Russia (Fig. 1B).Despite the absence of complete VP1 nt sequences, partial VP1 sequences (600 nucleotides) of the GII.P2-GII.2 strains from Russia in the sub-lineage 2 of lineage 1 were also found to be most related to those of BJFTJYX, BJFTZTX, and BJFTNJY, also belonging to the emerging GII.P2-GII.2 norovirus.This suggests that the emerging GII.P2-GII.2 strain in China may have been introduced from the strains in Russia and that the newly emerged viruses had spread across the countries.The strains in the sub-lineage 2 of lineage 1 were revealed to be relatively closely related to the 2009-2011 GII.P2- GII.2 strains, including the Rotterdam-5-709/NL/2011 (KX446510.1) and Rotterdam-5-0/NL/2009 (KX446499.1) strains.The MRCA of the emerging GII.P2-GII.2 strain appeared in 2012, which is consistent with the results of the analysis of the GII.2 VP1 genes (Fig. 1B).Hence, we conclude that the GII.P2-GII.2 strain emerging in 2016 is a novel recombinant virus that was generated from the 2011-2012 GII.P16-GII.2 and 2011 GII.P2-GII.2 strains in approximately 2011-2012.
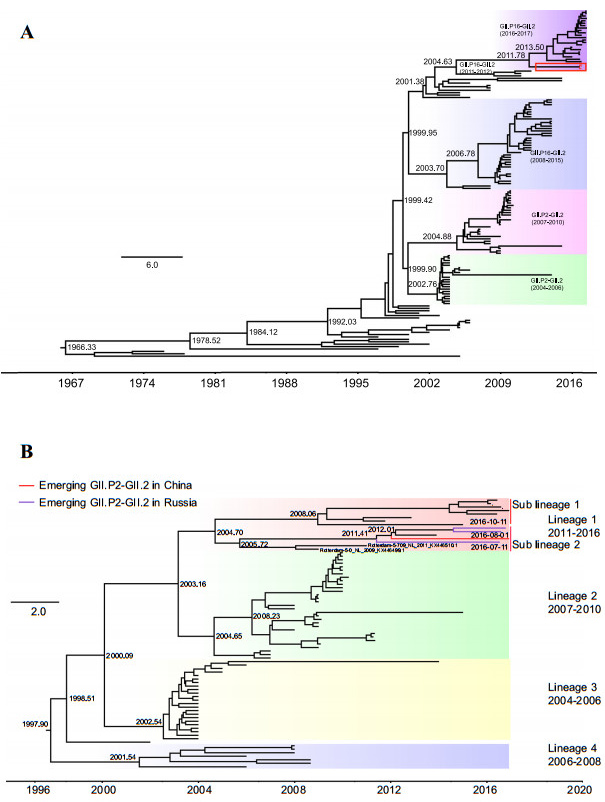
Figure 1. Maximum clade credibility (MCC) Bayesian phylogenetic trees of the GII.P2-GII.2 strains. (A) MCC tree of the VP1 genes of the GII.2 strains. The tree was constructed using 148 VP1 gene sequences of GII.2 strains using the GIR + G nucleotide substitution, strict clock, and Bayesian skyline model, as the tree prior. The three emerging GII.P2–GII.2 strains identified in the Fengtai District, Beijing City, China, in December 2016 are outlined by a red square frame. (B) The MCC tree of partial RdRp genes from GII.P2 strains. It was constructed using 79 GII.P2 partial RdRp sequences (720 nucleotides) using the HKY? G nucleotide substitution, strict clock, and Bayesian skyline models. The emerging GII.P2–GII.2 strains in China and Russia are indicated by red and purple branches, respectively. The phylogenetic clusters in the two trees are shown using different colors and names. Estimated divergence times of the common ancestor are shown at the key nodes. Scale bar, actual time (years).
The complete genome sequences of the reemerging GII.P16-GII.2 strain BJSMQ (KY421122.1) and GII.P2- GII.2 strain Hokkaido-16(LC209465.1) were used as reference sequences to further predict the recombination nucleotide breakpoint of the emerging GII.P2-GII.2 strain BJFTJYX by the Simplot software v.3.5.1.The SimPlot analysis showed that BJFTJYX shared the highest nucleotide sequence identity with the ORF1 of the GII.P2- GII.2 strain HenrytonSP17, and with the ORF2 and ORF3 of the reemerging GII.P16-GII.2 strain BJSMQ (Supplementary Figure S1).The recombination breakpoint was predicted to be at nucleotide 5177, near the ORF1/ORF2 overlap region (Supplementary Figure S1).
Pairwise comparison of all GII.2 VP1 sequences over 4 decades showed that the GII.2 prototype Snow Mountain Virus (SMV)(AY134748.1) showed an aa divergence of <3%, although the root-to-tip divergence plot of the VP1 nt sequences showed a strong clock-like evolution with a coefficient of determination (R2) value of 0.96.The analysis of VP1 sequences revealed that the emerging GII.P2- GII.2 strains showed maximum nt identities of approximately 99% to the 2016-2017 reemerging GII.P16-GII.2 strain CQ25(KY421149.1), while they showed the highest aa identity to the GII.P2-GII.2 strain OC08079 (BAL60765.1), with a divergence of only 1 aa.The alignment of GII.2 VP1 sequences also showed that although the residues adjacent to the HBGA-binding site I appeared to change over time compared to SMV, the major residues of three sites constituting the HBGA-binding interface remained conserved in the emerging GII.P2-GII.2 strain.These data indicate that GII.2 noroviruses have remained very stable over 4 decades, supporting the model presented by Parra et al.(2017).RdRp sequence analysis revealed that the RdRp proteins from these GII.P2 strains showed an aa divergence of less than 1%, although the root-to-tip divergence plot of the GII.P2 partial RdRp nt sequences revealed a moderate clock-like evolution over time (R2=0.60).The RdRp gene of the emerging GII.P2- GII.2 strain BJFTJYX shared 93.5%-95% nt and 99.0%- 99.6% aa identities with those of the previous GII.P2-GII.2 strains.The sequence analyses indicate that the RdRp proteins from GII.P2 strains are highly stable at the aa level.
Our analyses suggest that the GII.P2-GII.2 strain that emerged in 2016 harbors a capsid sequence identical to that in the reemerging GII.P16-GII.2 strain.However, the reemerging GII.P16-GII.2 strain, but not the emerging GII.P2-GII.2 strain, became predominant in 2016-2017 in Fengtai, implying that factors other than capsid proteins contribute to the reemerging GII.P16-GII.2 virus epidemic (Tohma et al.2017; Ao et al.2018).Epidemiological data also found that the GII.P2-GII.2 strains were identified during 1989-2010 and were gradually replaced by the GII.P16-GII.2 strains, which started to circulate in 2008 (Mizukoshi et al.2017).Thus, a possible explanation for the predominance of the reemerging GII.P16-GII.2 viruses is that their GII.P16 polymerase could have a bigger impact on fitness than the GII.P2 polymerase.Enhanced global surveillance of GII.2 genotype norovirus epidemiology is necessary to further understand its evolution and significance in public health.
HTML
-
This work was supported by the Special National Project on Research and Development of Key Biosafety Technologies (2016YFC120 1900) and the National Natural Science Foundation of China (31500139).
-
The authors declare that they have no conflict of interest.
-
This study was approved by the Institutional Review Board of the China CDC for human subject protection.
Conflict of interest
Animal and Human Rights Statement
-
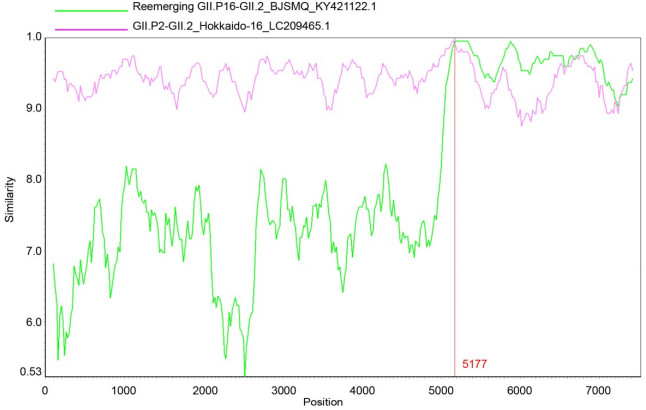
Figure S1. SimPlot analysis of the complete genomic sequence of the emerging GII.P2/GII.2 strain for recombination. SimPlot analysis was reconstructed with a slide window width of 200 bp and a step size of 20 bp. The query sequence of the emerging GII.P2-GII.2 strain BJFTJYX was aligned with the two reference strains as shown at the windows. The X-axis indicates the nucleotide positions of the NoV sequences, while the Y-axis indicates nucleotide similarity (%) between the emerging GII.P2-GII.2 strain and the reference strains.

 DownLoad:
DownLoad: