












HTML
-
Enterovirus species A71 (EV-A71), a member of the Enterovirus genus of the Picornaviridae family, is known to cause hand, foot, and mouth disease (HFMD) in children < 5 years of age (Solomon et al. 2010; Huang et al. 2018). EV-A71 associated HFMD typically presents as a selflimiting illness. However, some patients rapidly develop neurological and systemic complications, such as acute flaccid paralysis, brainstem encephalitis, cardiopulmonary dysfunction, neurological pulmonary edema and shock (Huang et al. 1999; Ooi et al. 2010; Lee et al. 2018). Since 2008, nearly 2 million cases have been reported in China every year, making the disease a serious public health concern (Li et al. 2018; Yu and Cowling 2019). The severity of the disease is increased by the fact that children younger than 5 years are especially susceptible to EV-A71 infection and the emerging epidemics collectively indicate that EV-A71 has efficient mechanisms for evading host's antiviral innate immunity. To date, few established antiviral treatments are available for EV-A71-induced severe HFMD (Yu and Cowling 2019).
EV-A71 was first isolated and described in 1969 (Schmidt et al. 1974). The non-enveloped, icosahedral RNA virus particle harbors a single-stranded RNA genome of approximately 7.4 kb in size, which encodes four structural viral proteins (VP1–VP4) and seven non-structural proteins (2A–2C and 3A–3D) with cis-cleavage sites of viral 2A protease (2Apro) and 3C protease (3Cpro) (Plevka et al. 2012; Wang and Li 2019). 2Apro has been classified as a cysteine protease with three critical amino acid residues comprising the catalytic core (H21-D39-C110) (Yuan et al. 2018). Apart from the essential role in the maturation of viral proteins, 2Apro also cleaves or reduces several cellular proteins mainly related to host cell cap-dependent translation and antiviral innate immunity (Zhang et al. 2018).
The process of cap-dependent translation requires eukaryotic initiation factor 4GI (eIF4GI), a large scaffolding protein bearing the docking sites for eIF4E (a capbinding protein) and eIF4A (an RNA helicase). The central role of eIF4GI in translation initiation makes it a valid target for modulating the efficiency of cap-dependent translation. For example, viral 2Apro from poliovirus and EV-A71 hydrolyzes eIF4GI at positions 674/681, which results in a rapid shutdown of cap-dependent translation (Hsu et al. 2009; Castello et al. 2011). In addition, during the onset of apoptosis, caspase-3-mediated proteolysis of eIF4GI at positions 532/1175 also causes translational suppression (Castello et al. 2011). Despite the fact that EVA71 is proved to induce apoptosis (Wang et al. 2015a, b), the contribution of caspase-3 in EV-A71-mediated eIF4GI cleavage has not yet been determined.
Host antiviral innate immunity, the first barrier against viral infections, is characterized by interferon (IFN) production and followed by IFN signaling. After infection by an RNA virus, IFN-α and -β production are induced upon sensing viral RNA by toll-like receptors (TLR) and RIG-Ilike receptors (RLR). Newly synthesized IFN-α or -β is secreted from cells and attaches to interferon alpha receptor (IFNAR) complex on the cellular membrane, thereby initiating the downstream IFN signaling pathway. Subsequently, Janus kinase (JAK) is activated, which results in the activation and phosphorylation of the signal transducer and activator of transcription (STAT). The phosphorylated STAT then interacts with IFN-regulatory factor 9 (IRF9) to form the interferon-stimulated gene factor 3 (ISGF3) complex. Finally, Karyopherin a1 (KPNA1) facilitates ISGF3 translocation to the nucleus, which activates the interferon-stimulated response element (ISRE), driving the expression of different ISGs (ISG56, MxA, PKR, etc.) (Chow and Gale 2015).
ISGs induced by IFN-α or -β act as antiviral proteins, which mainly disturb viral protein synthesis, thus inhibiting the production of viral progeny (Sadler and Williams 2008). However, the successful infection and proliferation of EV-A71 indicates an ability to avoid the above mentioned innate immune system in both phases of IFN production and IFN signaling. Indeed, viral 2Apro and 3Cpro are known to block IFN production by cleaving or binding the key upstream signal molecules (Jin et al. 2018). Additionally, the collapse of IFN-IFNAR1-JAK-STAT pathway especially the reduction of IFNAR1, caused by viral 2Apro, has been shown to be correlated with poor responsiveness to exogenous IFN treatments (Rasti et al. 2019; Morrison and Racaniello 2009). Finally, cellular caspases are also reported to promote disturbance of IFN production and IFN signaling in EV-A71 infected cells (Kuo et al. 2013; Wang et al. 2017). Altogether, the mechanism by which EV-A71 escapes the innate immune response is predictably multisided and complicated, implicating a virus-host interaction. In the present study, we focused on how viral and host cell key proteins downregulate IFNAR1 protein at the translation level, and we hoped to identify a potential antiviral-strategy for promoting the clinical application of IFN-α with less side effect.
-
Human rhabdomyosarcoma (RD) cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco) containing 10% fetal bovine serum (FBS, Hyclone) with 100 U/mL penicillin and 100 μ/mL streptomycin. EVA71 (BC08 strain, GenBank accession number JQ514785.1) was propagated and titrated in RD cells and stored at - 80 ℃ until use.
-
RD cells were infected with EV-A71 at the multiplicity of infection (MOI) indicated in the figure legends. For IFN stimulation or treatment, different concentrations of human IFN-α (#ab73124, Abcam) were used at the time points indicated in the figure legends. Puromycin (#P8230, Solarbio) was applied for inducing apoptosis. Inhibitor of MEK pathway (U0126, #PHZ1283, Thermo Scientific) and inhibitor of caspase-3 activation (Z-DEVD-FMK, #14414, Cayman) were also used as required.
-
To construct human IFNAR1 expression plasmid, a fulllength human IFNAR1 cDNA was cloned from HEK293 mRNA into the modified pcDNA3.1 vector with a C-terminal HA-tag. Construction of an N-terminal GFPtagged EV-A71 2A expression plasmid and the 2A inactivation mutation of Cys110 to Ala110 (yielding 2AC110A), as well as the construction of bi-cistronic reporter plasmid containing Cap-Rluc-vIRES-Fluc and its transcription in vitro were done as previously described (Duan et al. 2017). Reporter plasmids pGM-ISRE-RLuc and pGM-Fluc were purchased from Yeasen Co., Ltd.
The siRNA targeting human caspase-3 (si#1 and si#2) and the negative control siRNA (si#NC) were synthesized by Genepharma Co., Ltd. The sequences are as follows: si#1 50-UGGAUUAUCCUGAGAUGGGTT-30 (sense), 50- CCCAUCTCAGGAUAAUCCATT-30 (anti-sense); si#2 50-AGUGAAGCAAAUCAGAAACTT-30 (sense), 50- GUUUCUGAUUUGCUUCACUTT-30 (anti-sense); si#NC 50-UUCUCCGAACGUGUCACGUTT-30 (sense), 50- ACGUGACACGUUCGGAGAATT-30 (anti-sense). The above siRNA target sequences have been confirmed to be functional in a previous study (Wang et al. 2017).
The plasmids and siRNA transfection experiments were performed using Lipofectamine 2000 reagent (Life Technologies) according to the manufacturer's instruction.
-
Western blot was performed as previously described (Zhu et al. 2015; Zhang et al. 2010). Anti-EV-A71 VP1 antibody was obtained from Abnova (PAB7631-D01P, USA). Antibodies for eIF4GI (#2858), STAT1 (#9172), JAK1 (#3332), CASPASE-3 (#9662), PARP (#9542), phosphorylated STAT1 (#9167) and JAK1 (#3331) were purchased from Cell Signaling Technology. The endogenous IFNAR1 was detected with a specific antibody from Abcam (ab45172). Overexpression of GFP or HA tagged plasmids were detected with anti-GFP (#BE 2070) or anti-HA (#BE 2071) purchased from EASYBIO. These target proteins were detected with anti-rabbit or mouse secondary antibody conjugated with horseradish peroxidase (#BF03008 and #BF03001, Biodragon). Specific bands were visualized with enhanced chemiluminescent substrate (ECL). Each blot was stripped and reblotted with anti-β-actin or GAPDH (Sigma) for loading control. Each immunoblot assay was carried out at least three times and one of them was presented.
-
Total RNA from Vector or 2A overexpressed cells were isolated with TRIzol reagent (Invitrogen). About 1 μ total RNA were reverse transcribed into cDNA using a reverse transcription system (Invitrogen). Quantitative reverse transcription-PCR (qRT-PCR) was performed using SYBR GoTaq qPCR Master Mix (Promega) on an ABI Fast 7500 real-time system instrument. Amplification of IFNAR1 and β-actin mRNA were both conducted under the following thermal cycling condition: 95 ℃ for 10 min, followed by 40 cycles of 95 ℃ for 15 s and 60 ℃ for 1 min. Fluorescence signals were collected by the machine during the extension phase of each PCR cycle. The threshold cycle (CT) value was normalized to that of β-actin. Primers for human IFNAR1 (forward: 50-CACTTCTTCATGGTATGAGGTTG ACT-30, reverse: 50-ATTGCCTTATCTTCAGCTTCTAAATGT-30) and internal β-actin (forward: 50-ACTGTGCCCATCTACGAGG-30, reverse: 50-CAGGCAGCTCGTAGCTCTT-30) were used. All samples were run in triplicate, and the experiment was repeated three times. The relative mRNA level of IFNAR1 was expressed as fold change relative to the value of corresponding control (set as 1).
-
To monitor the effect of EV-A71 2Apro on the cap-dependent translation efficiency, bi-reporter mRNAs containing CapRluc-vIRES-Fluc were used in this study. RD cells were seeded in 96-well plates and transfected with bi-reporter mRNAs (100 ng/well) at indicated time points. Cells were then lysed for luciferase assay using the Luciferase Assay System Kit (Promega) according to manufacturer's instructions. Each luciferase reporter assay was carried out at least three times. Additionally, to measure the activity of ISRE, RD cells were first co-transfected with p-ISRE-FLuc (100 ng/well, 96-well plate) and p-RLuc (20 ng/well, 96-well plate) for 24 h. This was followed by re-treatment of the cells with EV-A71, 2A, DEVD or IFN-α as required. The intensities of Firefly luciferase (Fluc) and Renilla luciferase (Rluc) were measured as described above.
-
Data were analyzed using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA) and expressed as the mean ± standard deviation (SD) obtained from the experiments repeated at least three times. A P value < 0.05 level was considered statistically significant.
Cell Culture and Virus Preparation
Cell Infection, Stimulation and Inhibition
Plasmids, siRNA Oligos and Transfection
Western Blot and Antibodies
qRT-PCR
Luciferase Reporter Assay
Statistical Analysis
-
As shown in Fig. 1A, IFN-α post-treatment could not prevent the occurrence of the cytopathic effect (CPE) of RD cells induced by EV-A71 unlike with IFN-α pre-treatment. Accordingly, in comparison with mock-treatment, IFN-α post-treatment failed to decrease viral titers (TCID50/mL) in cell supernatants (P > 0.05) (Fig. 1B). Furthermore, with accumulation of VP1 in cells, the levels of membrane and total IFNAR1 as well as the phosphorylation of JAK1 and STAT1 were dramatically down-regulated (Fig. 1C). As we know, ISRE, downstream of IFNAR1-JAK-STAT pathway (Chow and Gale 2015), is a cis-acting element responsible for the expression of antiviral proteins (ISG56, Mx1, PKR, etc.). Our ISRE luciferase assay showed that the suppression of ISRE activities was associated with the down-regulation of IFNAR1 protein (Fig. 1D). Collectively, viral resistance to IFN-α signaling by reducing IFNAR1 protein levels is an important strategy for EV-A71 proliferation.
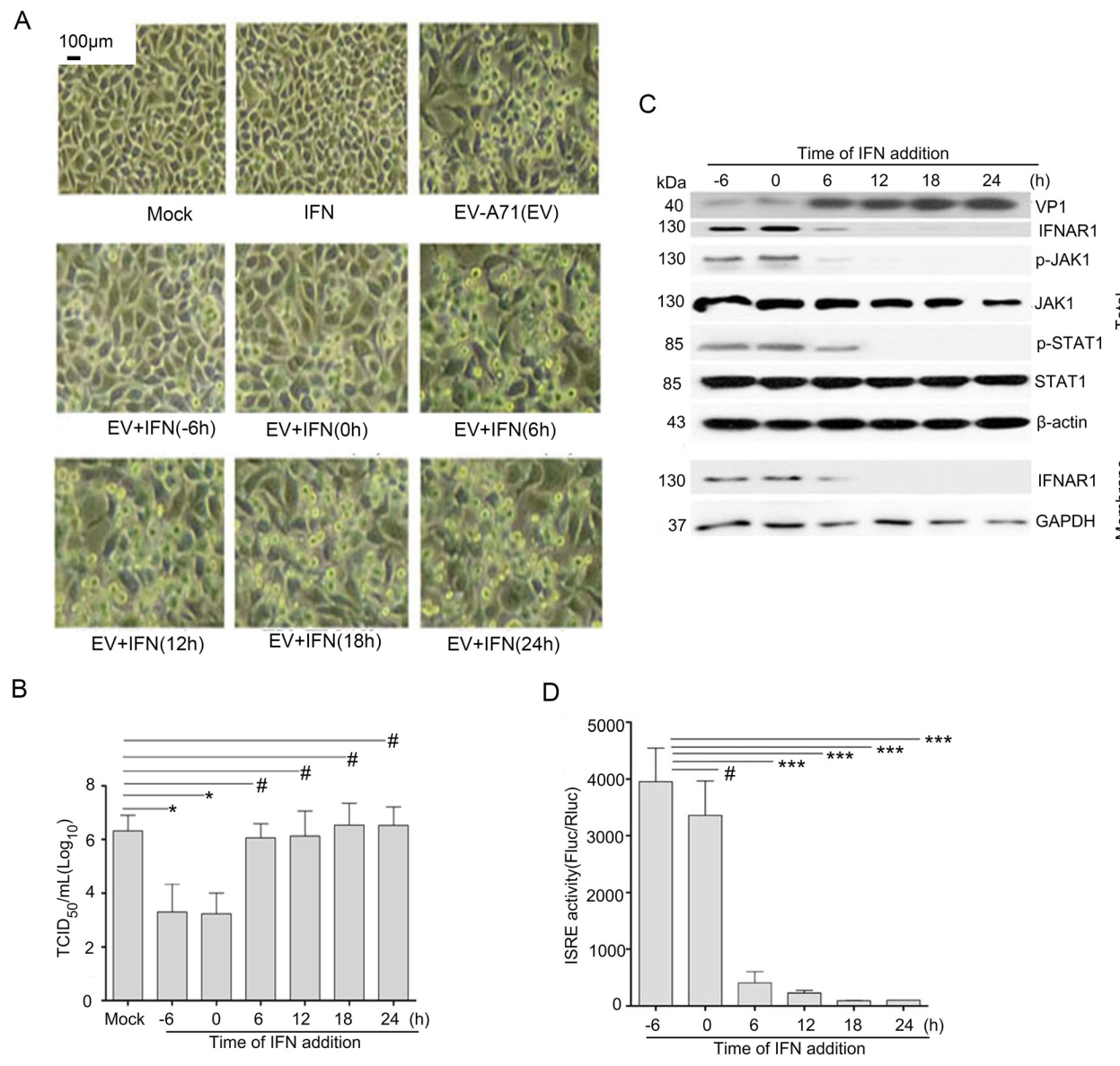
Figure 1. The effect of EV-A71 infection on IFN-α treatment. A RD cells were infected with EV-A71 (BC08 strain, Accession Number: JQ514785.1) at an MOI of two. IFN-α (1000 IU/mL) treatment was performed at 6 h before infection (-6 h), at the time of infection (0 h), and at 6 h, 12 h, 18 h, 24 h post-infection (p.i.), respectively. Photomicrographs were taken at 24 h p.i. (original magnification, 100 9). B Progeny viruses in supernatant were titrated using RD cells and the results (log10 TCID50/mL) indicate the mean ± SD of three independent experiments. Statistical analysis was performed using Student's t test, #P>0.05, *P < 0.05. C RD cells were treated in parallel as shown in (A), total cellular protein and membrane protein were extracted as described in MMs. The quantity of the indicated proteins in cell lysates was measured by Western blot. β-actin in total proteins and GAPDH in membrane proteins were used as loading control. D RD cells were firstly co-transfected with p-ISRE-FLuc (100 ng/well, 96-well plate) and p-RLuc (20 ng/well, 96-well plate) for 24 h, then the cells were treated in parallel as shown in (A). Results (Fluc/Rluc) indicate the mean ± SD of three independent experiments. Statistical analysis was performed using Student's t-test, #P>0.05, ***P < 0.001.
-
In order to verify the down-regulation mechanism of IFNAR1 protein, we monitored the IFNAR1 protein level in mock- or IFN-α-treated RD cells, since IFNAR1 protein was reported to have a lower expression level in a variety of tumor cells (Araya and Goldszmid 2017; Fuchs 2013). As shown in Fig. 2A, IFNAR1 protein levels were significantly increased in the cells treated with IFN-α, compared with the mock cells, suggesting an IFN-α-induced up-regulation of IFNAR1 translation in a cap-dependent manner. It is known that enteroviral 2Apro is a key viral protease responsible for the shutdown of cellular cap-dependent translation (Lee et al. 2017), which was also confirmed in the cells transfected with Cap-Rluc-vIRES-Fluc mRNA reporter in this study (Fig. 2B). Notably, overexpression of 2Apro had no significant inhibiting effect on IFN-α-induced IFNAR1 mRNA (Fig. 2C). These data show that 2Apro mediates a cap-dependent translation shutdown without disturbing IFNAR1 mRNA level induced by IFN-α.

Figure 2. The effect of EV-A71 2Apro on IFN-α induced IFNAR1 mRNA level. A RD cells were treated with IFN-α (100 IU/mL or 1000 IU/mL) for 1 h, 6 h, 12 h and 24 h, respectively. The cell lysates were collected for measuring protein levels of IFNAR1 and β-actin by Western blot. B RD cells were pre-transfected with p-EGFPVec (Vector), p-EGFP-2A or p-EGFP-2AC110A, respectively. Subsequently, 12 h later, the cells were re-transfected with Cap-RlucvIRES-Fluc mRNA (100 ng/well, 96-well plate) for another 12 h. The intensities of Fluc and Rluc were detected as described in MMs. Results show the ratio of Rluc intensity to Fluc intensity. The results (Rluc/Fluc) indicate the mean ± SD of three independent experiments. Statistical analysis was performed by Student's t-test, #P>0.05, ***P < 0.001. C RD cells were transfected with p-EGFP-Vec (2 μg/well, 6-well plate) or p-EGFP-2A (2 μg/well, 6-well plate) for 24 h, then the cells were treated with or without IFN-α (1000 IU/mL) for another 2 h. Extraction of total RNA and quantification of IFNAR1 mRNA were performed as described in MMs. The mRNA level of β-actin was used for normalization. Statistical analysis was conducted using Student's t-test, #P>0.05, ***P < 0.001.
-
Cap-dependent translation requires intact eIF4GI (Prévôt et al. 2003). We measured the protein levels of IFNAR1 and eIF4GI in 2Apro overexpressed cells. As shown in Fig. 3A, the wild-type 2Apro efficiently cleaved eIF4GI and suppressed the up-regulation of IFNAR1 protein compared with the inactivated mutant 2AC110A. Application of U0126, a MEK specific inhibitor, attenuated 2Apro-mediated cleavage of eIF4GI, and the IFNAR1 protein level was also restored in RD cells (Fig. 3B). Co-transfection of RD cells with CMV-initiated IFNAR1 plasmid and 2A plasmid further verified the effect of viral 2Apro on IFANR1 protein (Fig. 3C). These data confirm that 2Apro-mediated cleavage of eIF4GI blocks the process of IFNAR1 translation relying on 2A's catalytic activity.
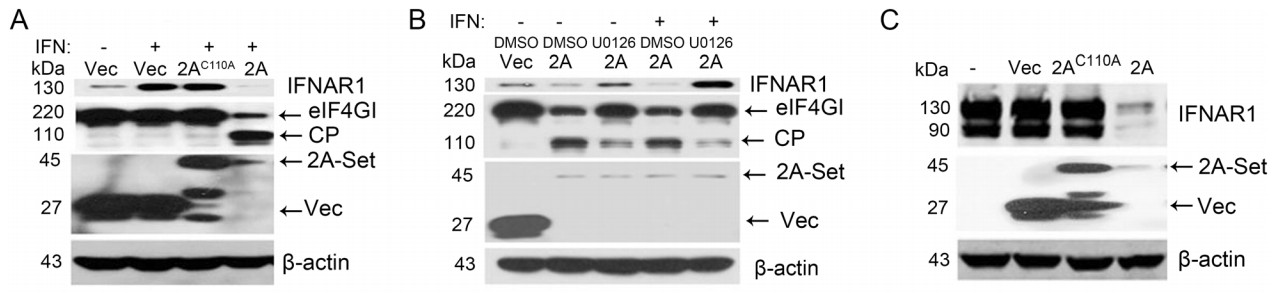
Figure 3. The effect of EV-A71 2Apro on IFNAR1 protein translation. A RD cells were treated in parallel as shown in Fig. 2C, then the cells were lysed with RIPA buffer for the detection of IFNAR1, eIF4GI, 2Apro or 2AC110A (2A-Set), Vector (Vec) and β-actin protein levels. CP, cleavage products. B RD cells were transfected with p-EGFP-Vec (2 μg/well, 6-well plate) or p-EGFP-2A (2 μg/well, 6-well plate) for 24 h, followed by mock treatment or IFN-α (1000 IU/mL) treatment for another 2 h. At 6 h prior to transfection, the cells were treated with U0126 (30 μmol/L) or its solvent DMSO (0.15%). Cell lysates were used to measure the indicated proteins by Western blot. C p-HAIFNAR1 (1.5 μg/well, 6-well plate) in combination with p-EGFP-2A (1.5 μg/well, 6-well plate) or p-EGFP-2AC110A (1.5 μg/well, 6-well plate) were co-transfected into RD cells for 36 h. Levels of the indicated proteins were measured by Western blot from the lysates with anti-HA and anti-GFP.
-
It has also been reported that eIF4GI serves as substrate for cellular caspase-3 in apoptotic cells (Marissen and Lloyd 1998). Based on reported data (Castello et al. 2011), the schematic diagram of eIF4GI and its cleavage sites by cellular caspase-3 and enteroviral 2Apro were summarized as showed in Fig. 4A. Translational inhibition was further measured after exposure of RD cells to puromycin, a caspase-3 activator. As shown in Fig. 4B, puromycin induced eIF4GI cleavage and arrested IFNAR1 translation (lane 4), which could be recovered by Z-DEVD-FMK (DEVD), the caspase-3 specific inhibitor (lane 5). Subsequently, the impact of 2Apro on caspase-3 activation was assessed. PARP cleavage, an indicator of caspase-3 activation, was observed in 2Apro over-expressed cells, suggesting that 2Apro was able to activate caspase-3 (Fig. 4C, lane 2). In addition, 2Apro cleaved eIF4GI, yielding 110 kDa and 80 kDa C-terminal truncated fragments (lane 2), but only the 80 kDa fragment bore the same molecular weight as that generated by caspase- 3 (lane 4). Interestingly, DEVD caused a reduction in both 110 kDa and 80 kDa fragments in 2Apro over-expressed cells (lane 3). The above data prove that caspase-3 is involved in 2Apro-mediated eIF4GI cleavage, which can lead to the translational arrest of IFANR1.
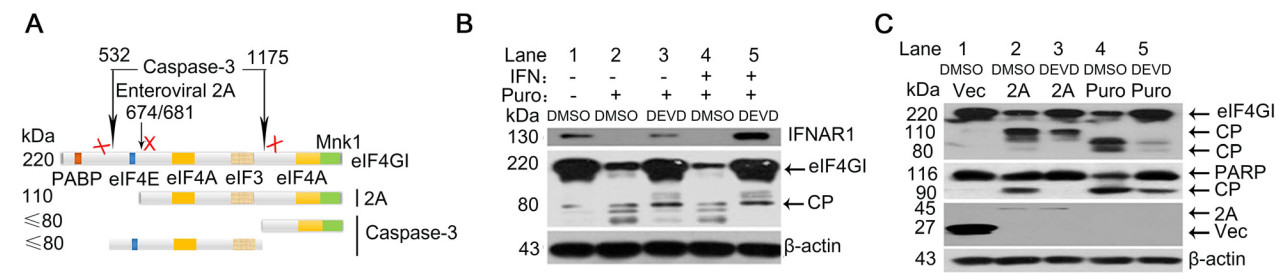
Figure 4. The effect of puromycin-activated caspase-3 on IFNAR1 protein translation. A Schematic diagram of eIF4GI and its cleavage sites. The boxes in eIF4GI indicate binding sites for PABP, eIF4E, eIF4A, elF3 and Mnk1 kinase, respectively. The black arrows indicate the cleavage sites of eIF4GI by cellular caspase-3 and enteroviral 2Apro, respectively. Cleavage of eIF4GI by 2Apro generates a nearly 110 kDa fragment. Caspase-3 directly cleaves eIF4GI and yields fragments, which are < 80 kDa in molecular weight. B RD cells were treated with puromycin (Puro, 10 μmol/L) for 6 h followed by mock treatment or IFN-α (1000 IU/mL) treatment for another 2 h. DEVD (20 mmol/L) or DMSO (0.2%) were pre-treated 6 h prior to puromycin treatment. Cell lysates were blotted on SDS-PAGE to detect the indicated proteins. CP, cleavage product. C RD cells were transfected with p-EGFP-2A (2 μg/well, 6-well plate) for 24 h or were treated with puromycin (10 μmol/L) for 6 h. DEVD (20 μmol/ L) or DMSO (0.2%) were pre-treated 6 h prior to 2A transfection or puromycin treatment. Cell lysates were used to determine the levels of indicated proteins. CP, cleavage product.
-
To reveal the possible role of caspase-3 in 2Apro-mediated translational suppression, we firstly observed the effect of DEVD on the up-regulation of IFNAR1 protein induced by IFN-α in 2Apro over-expressed cells. As shown in Fig. 5A, DEVD partially blocked cleavage of eIF4GI and increased IFNAR1 protein levels (lane 4) compared with the control (lane 3). Additionally, small interfering RNA (siRNA) targeting caspase-3 (si#1 and si#2) was further employed for confirming the above effect caused by DEVD. Results showed that the degree of eIF4GI cleavage were alleviated and IFNAR1 protein levels were partially recovered upon siRNA-mediated knockdown of caspase-3 expression (Fig. 5B, lane 3 and 4), in comparison with the control siRNA (lane 5). Above described data demonstrate that EVA71 2Apro-induced IFNAR1 translation arrest is partially mediated by caspase-3 activity.

Figure 5. The effect of caspase-3 inhibition on EV-A71 2Apro-mediated suppression of IFNAR1 protein translation. A RD cells were pretreated with DEVD (20 μmol/L, 6 h) or DMSO (0.20%, 6 h), and then p-EGFP-2A or p-EGFP-Vec (2 μg, 6-well plate) were transfected. At 4 h after transfection, the cells were cultured in medium with fresh DEVD or DMSO for 24 h and followed by IFN-α treatment for another 2 h. Cells were collected and lysed. Western blot was performed to analyze the levels of indicated proteins. B RD cells were transfected with caspase-3 specific siRNA-1 (si#1, 50 nmol/L), siRNA-2 (si#2, 50 nmol/L), or negative control siRNA (si#NC, 50 nmol/L), respectively. At 12 h after transfection, the cells were re-transfected with p-EGFP-Vec or p-EGFP-2A for another 24 h, followed by IFN-α treatment for another 2 h. Cell lysates were collected and analyzed using Western blot with specific antibodies to determine the protein levels of IFNAR1, eIF4GI, caspase-3 and GFPtagged 2A or Vec.
-
As caspase-3 activity is implicated in the process of 2Apro-mediated inhibition of IFNAR1 translation, caspase-3 inhibition would be highly beneficial for preserving IFN-α signaling. To test this hypothesis, the effect of DEVD on IFN-α-induced ISRE activity was firstly examined in 2Apro over-expressed cells. As observed with EV-A71 infection, ISRE activity was dramatically inhibited by 2Apro (Fig. 1D), but significantly restored by DEVD treatment (Fig. 6A). Subsequently, the effect of IFN-α in combination with DEVD on the proliferation of EV-A71 in vitro was further investigated. By observing the CPE caused by EV-A71, we found that a combination of DEVD and IFN-α treatment protected cells from CPE, compared with treatment by DEVD or IFN-α alone (Fig. 6B). In parallel, the co-treatment significantly inhibited VP1 expression and increased IFNAR1 protein levels. Notably, a very low concentration of IFN-α (200 IU/mL) was sufficient to inhibit replication of EV-A71 when combined with DEVD (Fig. 6C). Collectively, these results indicate that caspase-3 inhibition contributed to the activity of IFN-α against EVA71 proliferation by the restoration of IFNAR1 protein levels.
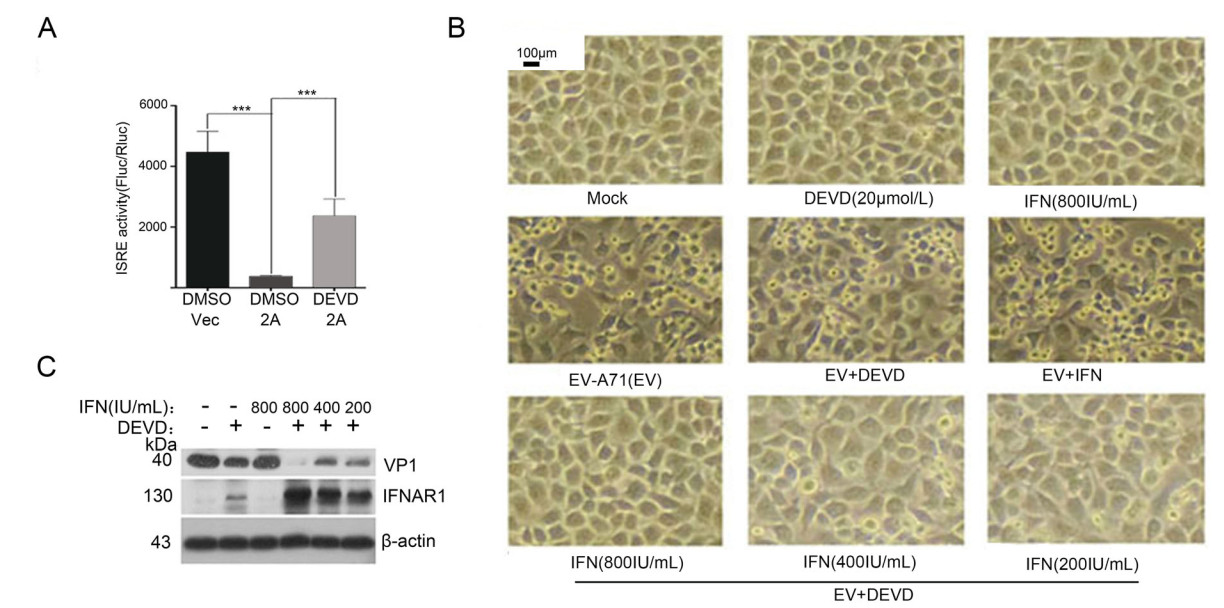
Figure 6. The effect of caspase-3 inhibitor plus IFN-α on the proliferation of EV-A71. A RD cells were first co-transfected with p-ISREFLuc and p-RLuc for 24 h, followed by treatment of the cells in parallel to Fig. 5A. The detection of luciferase and analysis as described in Fig. 1C, ***P < 0.001. B RD cells were infected with EV-A71 at an MOI of 2. 6 h later, the cells were treated with IFN-α at the concentration of 200 IU/mL, 400 IU/mL and 800 IU/mL, respectively. Additionally, 6 h prior to EV-A71 infection, the cells were pretreated with DEVD at the concentration of 20 μmol/L. At 24 h p.i., photomicrographs were taken (original magnification, 100 ×). C The protein levels of viral VP1 and IFNAR1 in cell lysates were measured by Western blot.
EV-A71 Antagonized Antiviral Activity of IFN-α by Reducing IFNAR1 Protein Level
EV-A71 2Apro Did Not Disturb the IFN-α Induced IFNAR1 mRNA Level
EV-A71 2Apro Inhibited IFNAR1 Translation by Cleavage of eIF4GI
Caspase-3-Mediated Cleavage of eIF4GI Impaired IFNAR1 Translation
Caspase-3 was Involved in EV-A71 2Apro-Mediated Inhibition of IFNAR1 Translation
Inhibition of Caspase-3 Contributed to the Activity of IFN-α Against EV-A71 Proliferation
-
It has been reported by different groups that EV-A71 impairs IFN-α signaling pathway by down-regulation of IFNAR1 and JAK1 protein levels (Lu et al. 2012; Wang et al. 2015a, b; Liu et al. 2014), degradation of KPNA1 protein (Wang et al. 2017), and cleavage of IRF9 and PKR proteins (Hung et al. 2011; Chang et al. 2017). Reported data suggests that complex mechanisms are involved in IFN-α antagonism during EV-A71 infection. The level of cellular IFNAR1 protein, a ''portal'' molecule that directly binds to IFN-α, bears the most importance among the IFNIFNAR1 signal pathway. However, the mechanism of IFNAR1 protein down-regulation upon EV-A71 infection is still unknown.
In the present study, we focused on IFNAR1 translation events in IFN-α antagonism during EV-A71 infection. We found a rapid and significant increase of IFNAR1 protein in RD cells only treated with IFN-α (Fig. 2A), indicating that synthesis of new IFNAR1 protein occurs in the cells infected with EV-A71, as also reported by Lu et al. (2012), who showed that EV-A71 infection activated type Ⅰ IFN production. However, the increased IFNAR1 protein induced by IFN-α was blocked by pre-infection with EV-A71 (Fig. 1C), providing a key mechanism for virusmediated down-regulation of IFNAR1 protein at translation level. It is known that enteroviral 2Apro-mediated proteolysis of eIF4GI plays a key role in the shutdown of cellular cap-dependent translation. We showed that co-transfection of RD cells with plasmids expressing 2Apro and IFNAR1 resulted in the cleavage of eIF4GI and down-regulation of over-expressed IFNAR1 simultaneously, as well as that 2Apro catalyzed eIF4GI cleavage and prevented the upregulation of endogenous IFNAR1 protein without an effect on IFNAR1 mRNA level. Our results support the hypothesis that viral 2Apro impairs IFNAR1 cap-dependent translation by cleavage of cellular eIF4GI. Given that the catalytic triad of 2Apro is highly conserved in enteroviruses, the aforementioned mechanism hijacked by EV-A71 may be a reasonable explanation as to why viral 2Apro is essential for enterovirus replication in IFN-α treated cells (Morrison and Racaniello 2009). Collectively, EV-A71 2Apro-mediated inhibition in cap-dependent translation was identified as an important reason for the down-regulation of IFNAR1 protein in virus-infected cells.
Moreover, we identified that cellular proteinase caspase-3 contributed to EV-A71 2Apro-mediated down-regulation of IFNAR1 translation, as implicated by the enabled activation of caspase-3 by EV-A71 2Apro, and inhibition of caspase-3 resulting in the attenuation of viral 2Apro-mediated eIF4GI cleavage, leading to partial restoration of IFNAR1 protein. Notably, we observed that viral 2Apro (Fig. 4C lane 2) and caspase-3 (Fig. 4C lane 4) shared a nearly 80 kDa fragment cleaved from eIF4GI, but the 110 kDa fragment only existed in the cells transfected with 2A. This further verified that the cleavage sites in eIF4GI targeted by viral 2Apro and caspase-3 were different (Zamora et al. 2002). However, caspase-3 inhibition caused the reduction of both 80 kDa and 110 kDa fragments of eIF4GI in 2Apro-overexpressed cells (Fig. 4C lane 3), suggesting that cellular caspase-3 contributed to EV-A71 2Apro-mediated hydrolysis of eIF4GI through direct cleavage (yielding 80 kDa fragment) and a synergistic effect with 2Apro cleavage in an undetermined manner(s) (yielding 110 kDa fragment). Deszcz et al. (2004) showed that caspase-3 inhibitor blocked the cleavage of eIF4GI by 2Apro of human rhinovirus and coxsackievirus B4. Therefore, the phenomenon of host cell proteases being hijacked by viral 2Apro may be ubiquitous in Enteroviruses.
As mentioned before, we demonstrated that caspase-3 contributed to EV-A71 2Apro-mediated suppression of IFNAR1 translation. Moreover, convincing evidence of efficient treatment of EV-A71 infection by the combination of IFN-α and DEVD was provided in this study (Fig. 6C), indicating that a low dose of IFN-α (200 IU/mL) could also achieve an obvious antiviral effect when caspase-3 activation was blocked. It is worth noting that an effective antiviral action of combinational therapy is likely to be multifactorial, among which the recovery of IFNAR1 protein was proven the most important one. In addition, we and others found that DEVD alone had an inhibitory effect on EV-A71 replication (Song et al. 2018), the precise mode of action is supposedly attributed to the suppression of viral IRES-dependent translation mediated by alleviative cleavage of eIF4GI upon caspase-3 inhibition (Lee et al. 2017).
As mentioned above, EV-A71 infection could trigger the intrinsic immune mechanism through IFN-α-IFNAR signal activation, whereas viral 2Apro antagonizes antiviral activity of IFN-α by reducing IFNAR1 protein in the manners as illustrated in Fig. 7, which eventually leads to a successful viral infection. Our work provides novel evidence that both EV-A71 2Apro and cellular caspase-3 contribute to the down-regulation of IFNAR1 at translation level by the cleavage of eIF4GI, especially highlighting the contribution of cellular caspase-3, thereby allowing EVA71 to subvert the "portal" molecule IFNAR1 in the IFNa-IFNAR signaling pathway and avoid the cell intrinsic innate immune function. This allowed us to identify a potential combinational antiviral-strategy for promoting IFN-α application with fewer clinical side effects.
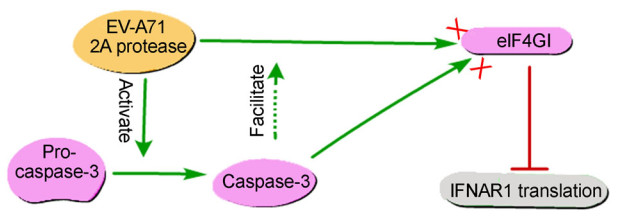
Figure 7. EV-A71 2Apro and cellular caspase-3 mediated down-regulation of IFNAR1 protein level at the translation level. EV-A71 2Apro-mediated cleavage of eIF4GI causes inhibition in IFNAR1 protein translation. Moreover, the caspase-3 activated by 2Apro can directly cleave eIF4GI and also facilitate 2Apro to cleave eIF4GI through an unknown mechanism.
-
This work was partially supported by grants from Beijing Natural Science Foundation (No. 19G10290) and National Natural Science Foundation of China (No. 81772184).
-
YP and HZ designed the experiments and analyzed the data. BC, YW and SW performed most of the experiments. BC wrote the manuscript. XP, HZ and YP revised the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no competing interests.
-
This study does not involve animal experiments.

 DownLoad:
DownLoad: