Hand, foot and mouth disease (HFMD) is a major public health concern in China. Since its first large outbreak in 2008, the dominant HFMD pathogens are constantly changing. In this study, Fu et al. performed meta-analyses on cohorts which have molecular epidemiological and enterovirus sequence data available in the public databases. They found that a rapid increase of other enteroviruses in accompany with a sharp decrease of EVA71 prevalence in China from 2008 to 2016, and four genotypes EVA71_C4, CVA16_B1, CVA6_D and CVA10_C contributed to over 90% of all HFMD cases in China. This study presents a comprehensive overview of the recent national epidemiology and evolutionary history of the major enteroviruses that cause HFMD in China. See page 22–34 for details.
Na Li, Zhiqiang Li, Yan Fu and Sheng Cao. Cryo-EM Studies of Virus-Antibody Immune Complexes[J]. Virologica Sinica, 2020, 35(1): 1-13. doi: 10.1007/s12250-019-00190-5.
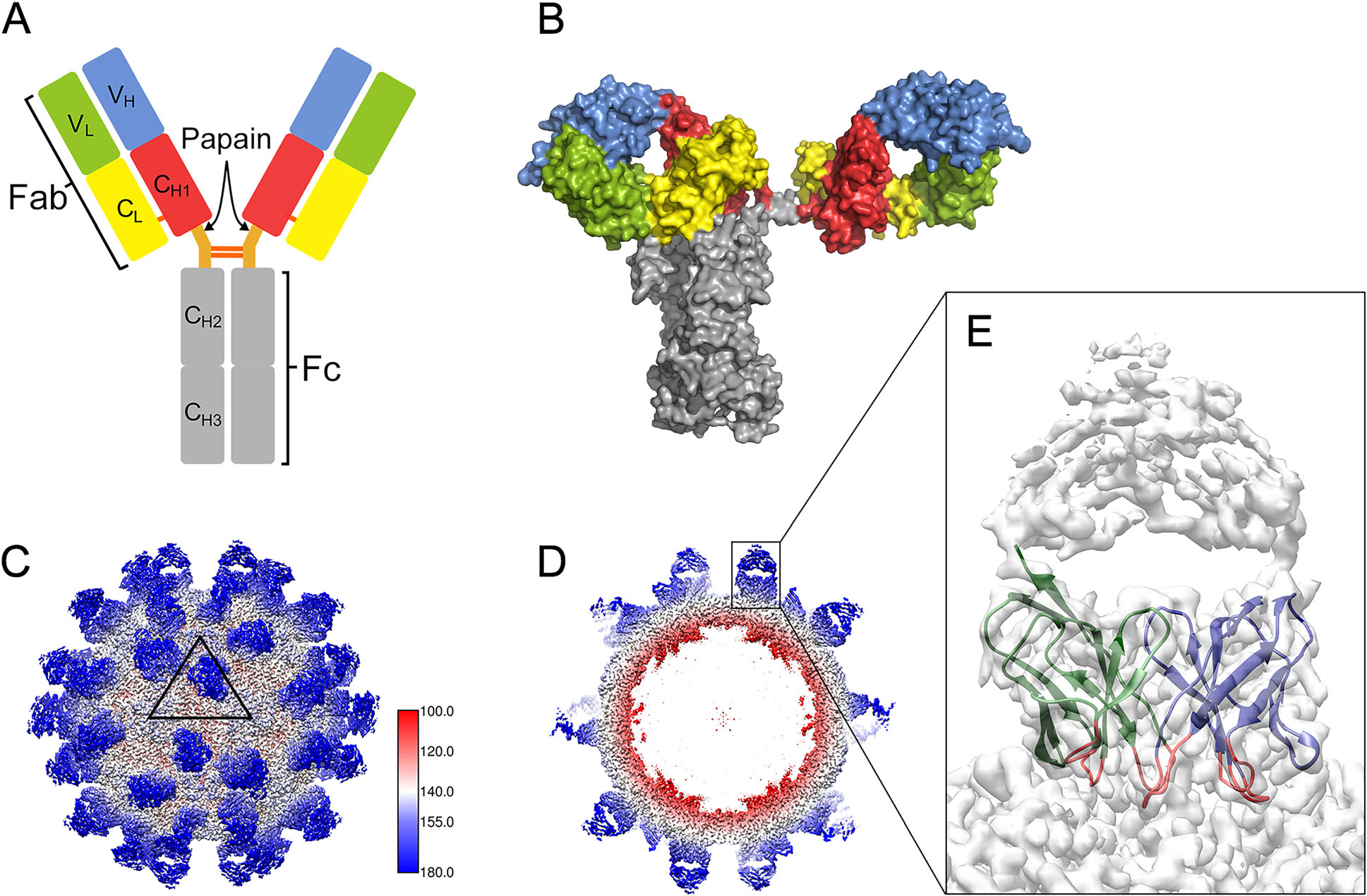
Antibodies play critical roles in neutralizing viral infections and are increasingly used as therapeutic drugs and diagnostic tools. Structural studies on virus-antibody immune complexes are important for better understanding the molecular mechanisms of antibody-mediated neutralization and also provide valuable information for structure-based vaccine design. Cryo-electron microscopy (cryo-EM) has recently matured as a powerful structural technique for studying bio-macromolecular complexes. When combined with X-ray crystallography, cryo-EM provides a routine approach for structurally characterizing the immune complexes formed between icosahedral viruses and their antibodies. In this review, recent advances in the structural understanding of virus-antibody interactions are outlined for whole virions with icosahedral T = pseudo 3 (picornaviruses) and T = 3 (flaviviruses) architectures, focusing on the dynamic nature of viral shells in different functional states. Glycoprotein complexes from pleomorphic enveloped viruses are also discussed as immune complex antigens. Improving our understanding of viral epitope structures using virus-based platforms would provide a fundamental road map for future vaccine development.
Xiaowen Li, Karen Kie Yan Chan, Bo Xu, Ming Lu and Bing Xu. Spatial, Temporal and Genetic Dynamics Characteristics of Influenza B Viruses in China, 1973–2018[J]. Virologica Sinica, 2020, 35(1): 14-20. doi: 10.1007/s12250-019-00161-w.
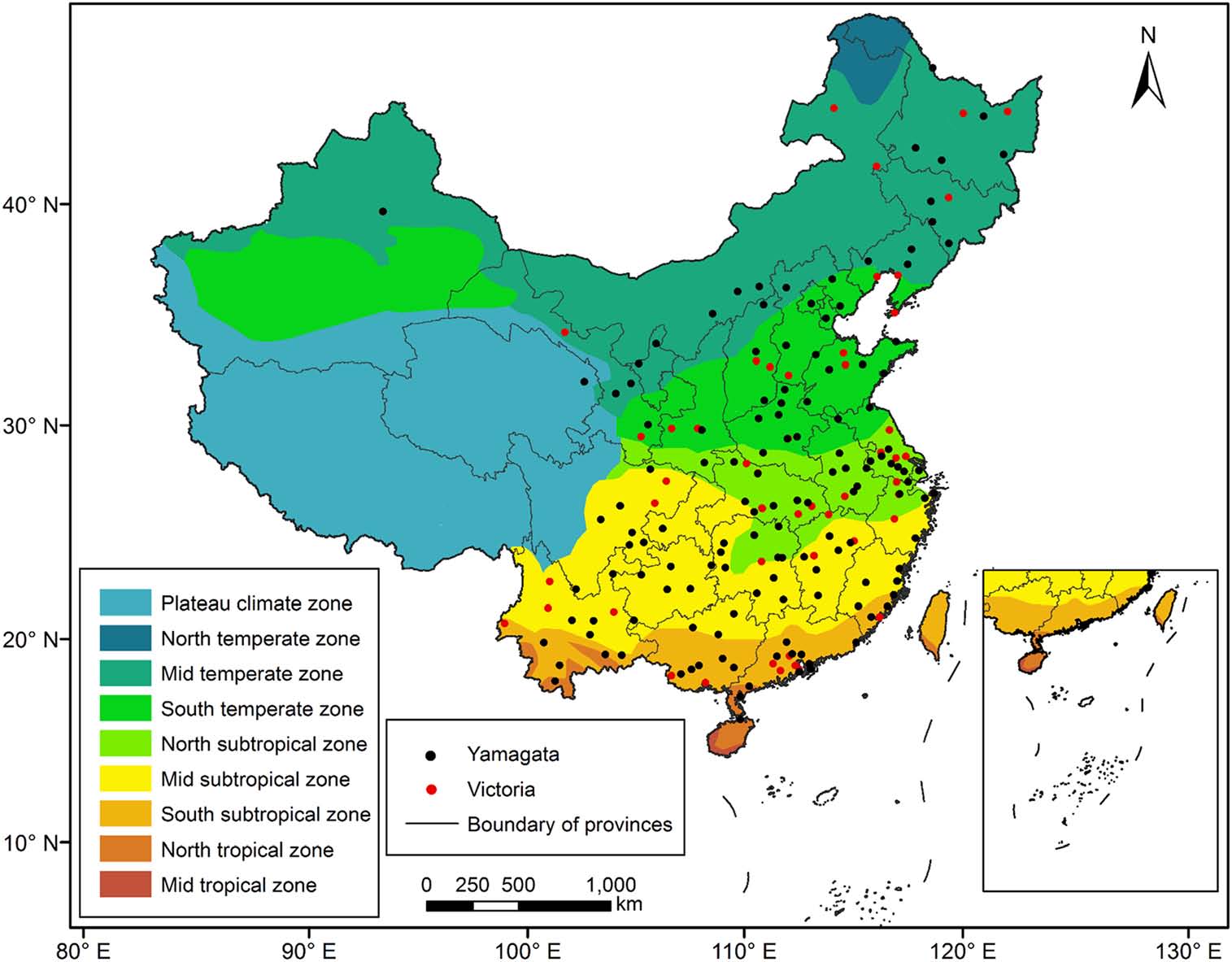
Annual influenza B virus epidemics and outbreaks cause severe influenza diseases in humans and pose a threat to public health. China is an important epidemic area of influenza B viruses. However, the spatial, temporal transmission pathways and the demography history of influenza B viruses in China remain unknown. We collected the haemagglutinin gene sequences sampled of influenza B virus in China between 1973 and 2018. A Bayesian Markov chain Monte Carlo phylogeographic discrete approach was used to infer the spatial and temporal phylodynamics of influenza B virus. The Bayesian phylogeographic analysis of influenza B viruses showed that the North subtropical and South subtropical zones are the origins of the Victoria and Yamagata lineage viruses, respectively. Furthermore, the South temperate and North subtropical zones acted as transition nodes in the Victoria lineage virus dispersion network and that the North subtropical and Mid subtropical zones acted as transition nodes in the Yamagata lineage virus dispersion network. Our findings contribute to the knowledge regarding the spatial and temporal patterns of influenza B virus outbreaks in China.
Xuemin Fu, Zhenzhou Wan, Yanpeng Li, Yihong Hu, Xia Jin and Chiyu Zhang. National Epidemiology and Evolutionary History of Four Hand, Foot and Mouth Disease-Related Enteroviruses in China from 2008 to 2016[J]. Virologica Sinica, 2020, 35(1): 21-33. doi: 10.1007/s12250-019-00169-2.
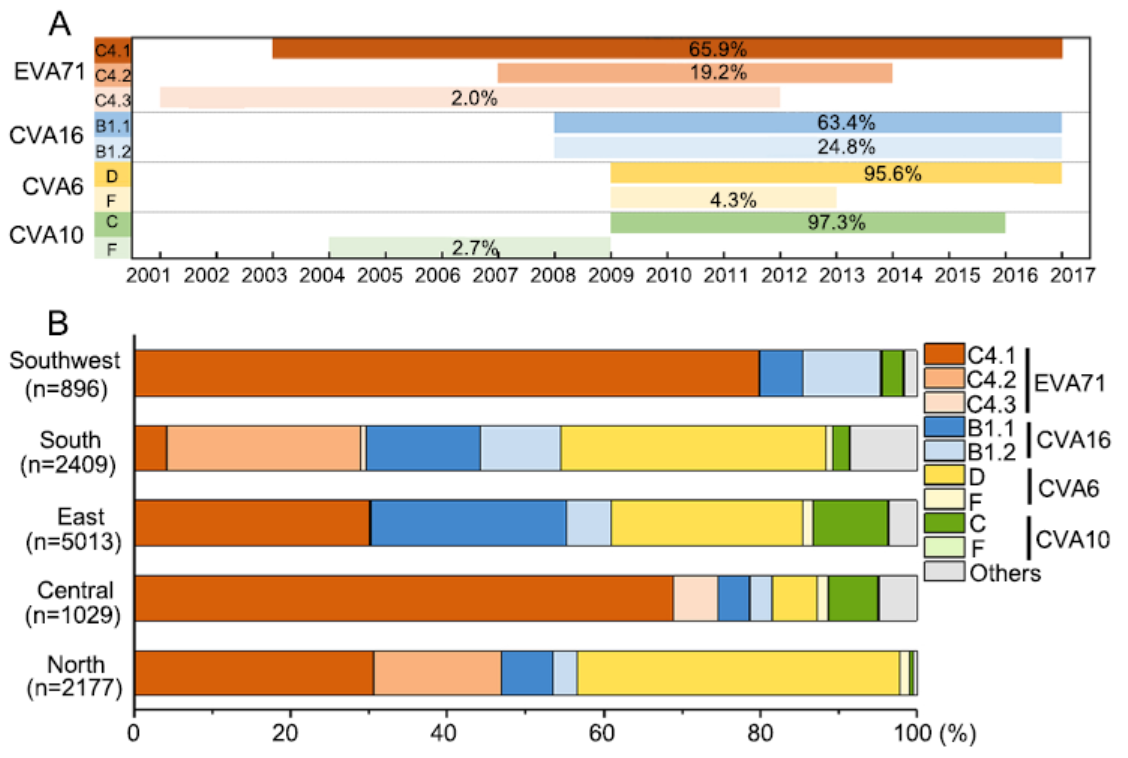
Hand, foot and mouth disease (HFMD) is a major public health concern in China. The most predominant enteroviruses that cause HFMD have traditionally been attributed to enterovirus A71 (EVA71) and coxsackievirus A16 (CVA16). Since its first large outbreak in 2008, the dominant HFMD pathogens are constantly changing. In 2013 and 2015, CVA6 exceeded both EVA71 and CVA16 to become the leading cause of HFMD in some provinces. However, there still lacks a comprehensive overview on the molecular epidemiology and evolution of HFMD-related enteroviruses at the national level. In this study, we performed systematic epidemiological analyses of HFMD-related enteroviruses using the data of 64 published papers that met the inclusion criteria, and conducted phylogenetic analyses based on 12, 080 partial VP1 sequences identified in China before 31st June 2018. We found that EVA71 prevalence has decreased sharply but other enteroviruses have increased rapidly from 2008 to 2016 and that one subtype of each enterovirus is represented during the epidemic. In addition, four genotypes EVA71_C4, CVA16_B1, CVA6_D and CVA10_C are the most predominant enterovirus strains and collectively they cause over 90% of all HFMD cases in China according to the phylogenetic trees using representative partial VP1 sequences. These four major enterovirus genotypes have different geographical distributions, and they may cocirculate with other genotypes and serotypes. These results suggest that more molecular epidemiological studies should be performed on several enteroviruses simultaneously, and such information should have implications for virological surveillance, disease management, vaccine development and policy-making on the prevention and control of HFMD.
Yuanyun Ao and Zhaojun Duan. Novel Primate Bocaparvovirus Species 3 Identified in Wild Macaca Mulatta in China[J]. Virologica Sinica, 2020, 35(1): 34-42. doi: 10.1007/s12250-019-00163-8.
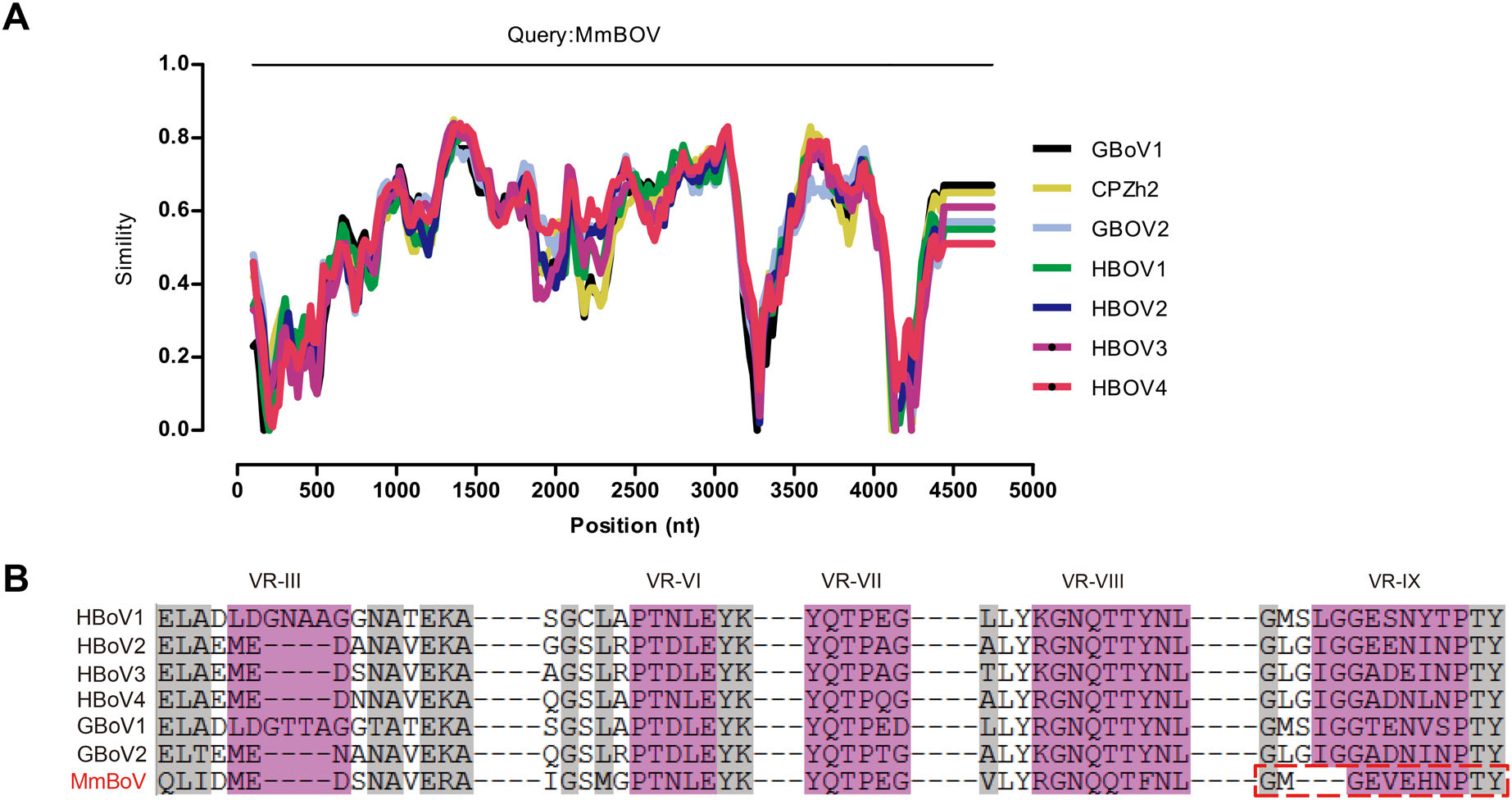
Primate bocaparvovirus (BOV) is a possible cause of respiratory disorders and gastroenteritis in humans. However, the diversity and evolution of these viruses remain largely unknown, despite the identification of a growing number of BOVs in non-human primates (NHPs). Here, we report the identification of a novel BOV (provisionally named Macaca mulatta bocaparvovirus [MmBOV]) in the feces of wild Macaca mulatta in China by viral metagenomic analysis. Seven of 400 fecal samples from Macaca mulatta were positive for MmBOV. An almost complete genome sequence of 4, 831 nucleotides was obtained, which had genomic organization and protein motifs similar to human bocaviruses (HOBVs), and shared characteristically low G/C content and weak codon usage bias. Sequence analyses of NS1, NP1, and VP1 revealed that MmBOV was most closely related to HBOV4 of Primate bocaparvovirus 2 (approximately 68.4%/70.6%, 73.3%/ 67.6%, and 70.4%/73.1% nucleotide/amino acid identities, respectively). Additionally, phylogenetic analysis revealed that MmBOV formed an independent peripheral branch, but clustered closely with those of the Primate bocaparvovirus species in the BOV genus (particularly HBOV4). These data strongly suggest that HBOV4 originated from NHP bocaparvoviruses around 200–300 years ago, and that NHPs may act as HBOV reservoirs. Following the International Committee of Taxonomy for Viruses guidelines, we propose MmBOV as a new species (tentatively named Primate bocaparvovirus 3) in the genus Bocaparvovirus, which is the first report of a novel species of primate BOV. Our data facilitate future research on the genetic diversity and evolution of primate bocaparvoviruses and highlight the importance of bocaparvovirus surveys in wild NHPs.
Yanmei Ma, Xiaoyong Chen, Keyuan Chen, Xiancheng Zeng, Shili Yang, Wei Chang, Yao Tang, Xiaoli Chen, Song Wang and Ji-Long Chen. Identification and Characterization of a Distinct Strain of Beak and Feather Disease Virus in Southeast China[J]. Virologica Sinica, 2020, 35(1): 43-51. doi: 10.1007/s12250-019-00159-4.
Beak and feather disease virus (BFDV) is an infectious agent responsible for feather degeneration and beak deformation in birds. In March 2017, an epidemic of psittacine beak and feather disease (PBFD) struck a farm in Fuzhou in the Fujian Province of southeast China, resulting in the death of 51 parrots. In this study, the disease was diagnosed and the pathogen was identified by PCR and whole genome sequencing. A distinct BFDV strain was identified and named as the FZ strain. This BFDV strain caused severe disease symptoms and pathological changes characteristic of typical PBFD in parrots, for example, loss of feathers and deformities of the beak and claws, and severe pathological changes in multiple organs of the infected birds. Phylogenetic analysis showed that the FZ strain was more closely related to the strain circulating in New Caledonia than the strains previously reported in China. Nucleotide homology between the FZ strain and other 43 strains of BFDV ranged from 80.0% to 92.0%. Blind passage experiment showed that this strain had limited replication capability in SPF Chicken Embryos and DF-1 Cells. Furthermore, the capsid (Cap) gene of this FZ strain was cloned into the pGEX-4T-1 expression vector to prepare the polyclonal anti-Cap antibody. Western blotting analysis using the anti-Cap antibody further confirmed that the diseased parrots were infected with BFDV. In this study, a PBFD and its pathogen was identified for the first time in Fujian Province of China, suggesting that future surveillance of BFDV should be performed.
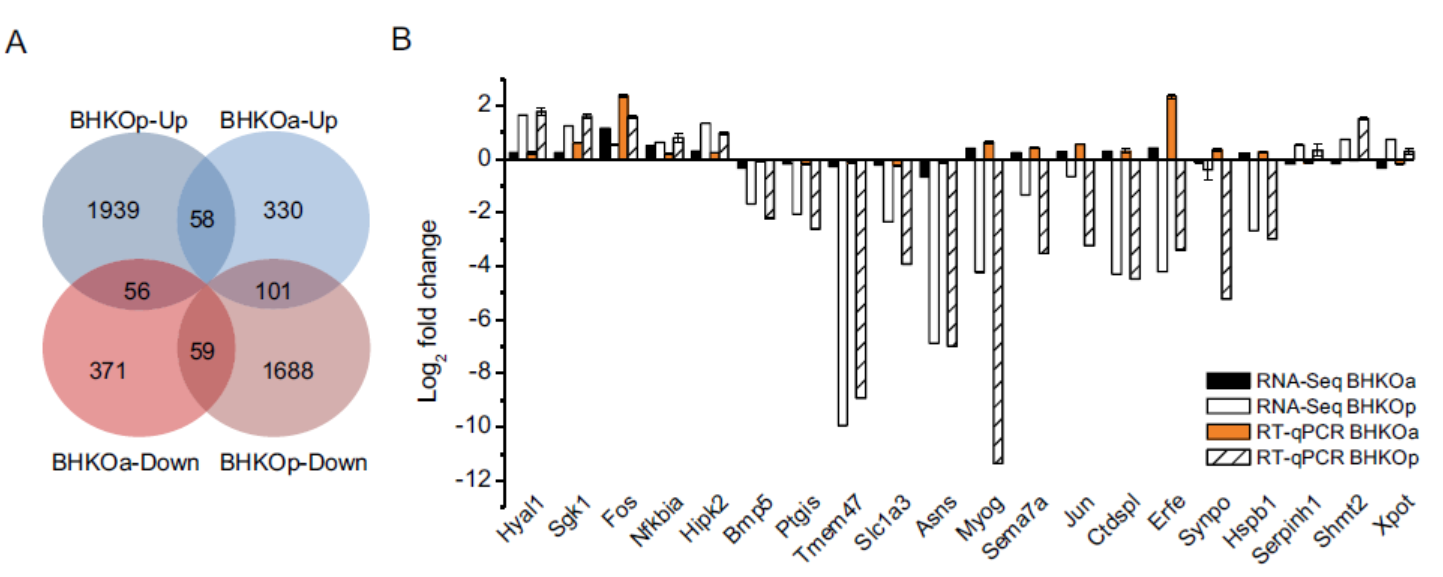
Jiadai Li, Lingling Han, Yao Hao, Yuncong Yuan, Mingzhen Wang, Xiu Xin, Hailong Wang, Fang Yu, Congyi Zheng and Chao Shen. Comparative Transcriptome Analysis Reveals Different Host Cell Responses to Acute and Persistent Foot-and-Mouth Disease Virus Infection[J]. Virologica Sinica, 2020, 35(1): 52-63. doi: 10.1007/s12250-019-00155-8.
Foot-and-mouth disease virus (FMDV) rapidly causes cytopathic effects in susceptible cells. Incomplete viral clearance during the acute infection leads to persistent infection. The relationship between host gene expression and the persistent infection remains unclear. In this study, we analyzed the transcriptome profiles of BHK-21 cells acutely and persistently infected with FMDV to identify differences in gene expression. GO and KEGG enrichment analyses showed that the 8, 378 differentially expressed genes were significantly enriched in categories including metabolism, biosynthesis, ribosome function, and endocytosis. In persistently infected BHK-21 cells, ribosome- and translation-related genes were significantly down-regulated. There were more differentially expressed immune-related genes during persistent infection than during acute infection. Two hundred and seventy-four genes were differentially expressed in both acutely and persistently infected BHK-21 cells. Among these genes, heat shock protein family B member 1 (Hspb1) knockdown significantly inhibited FMDV replication. Our research provides a basis for further research to understand the mechanisms of persistent FMDV infection including the genes involved in FMDV replication.
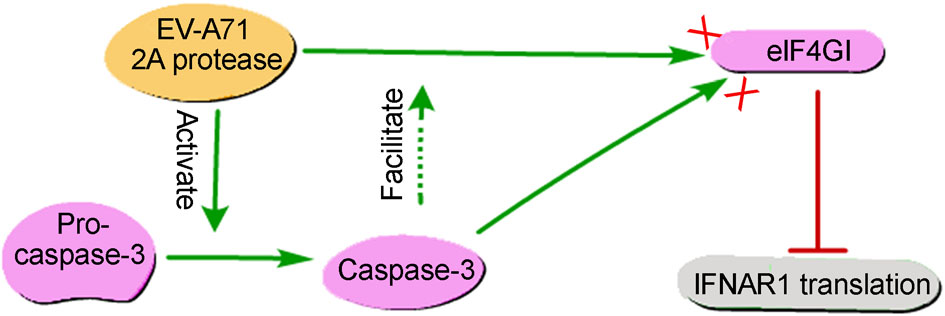
Bangtao Chen, Yuya Wang, Xinyi Pei, Sanyuan Wang, Hao Zhang and Yihong Peng. Cellular Caspase-3 Contributes to EV-A71 2Apro-Mediated DownRegulation of IFNAR1 at the Translation Level[J]. Virologica Sinica, 2020, 35(1): 64-72. doi: 10.1007/s12250-019-00151-y.
Enterovirus A71 (EV-A71) is the major pathogen responsible for the severe hand, foot and mouth disease worldwide, for which few effective antiviral drugs are presently available. Interferon-a (IFN-α) has been used in antiviral therapy for decades; it has been reported that EV-A71 antagonizes the antiviral activity of IFN-α based on viral 2Apro-mediated reduction of the interferon-alpha receptor 1 (IFNAR1); however, the mechanism remains unknown. Here, we showed a significant increase in IFNAR1 protein induced by IFN-α in RD cells, whereas EV-A71 infection caused obvious downregulation of the IFNAR1 protein and blockage of IFN-α signaling. Subsequently, we observed that EV-A71 2Apro inhibited IFNAR1 translation by cleavage of the eukaryotic initiation factor 4GI (eIF4GI), without affecting IFNAR1 mRNA levels induced by IFN-α. The inhibition of IFNAR1 translation also occurred in puromycin-induced apoptotic cells when caspase-3 cleaved eIF4GI. Importantly, we verified that 2Apro could activate cellular caspase-3, which was subsequently involved in eIF4GI cleavage mediated by 2Apro. Furthermore, inhibition of caspase-3 activation resulted in the partial restoration of IFNAR1 in cells transfected with 2A or infected with EV-A71, suggesting the pivotal role of both viral 2Apro and caspase-3 activation in the disturbance of IFN-α signaling. Collectively, we elucidate a novel mechanism by which cellular caspase-3 contributes to viral 2Apro-mediated down-regulation of IFNAR1 at the translation level during EV-A71 infection, indicating that caspase-3 inhibition could be a potential complementary strategy to improve clinical anti-EV-A71 therapy with IFN-α.
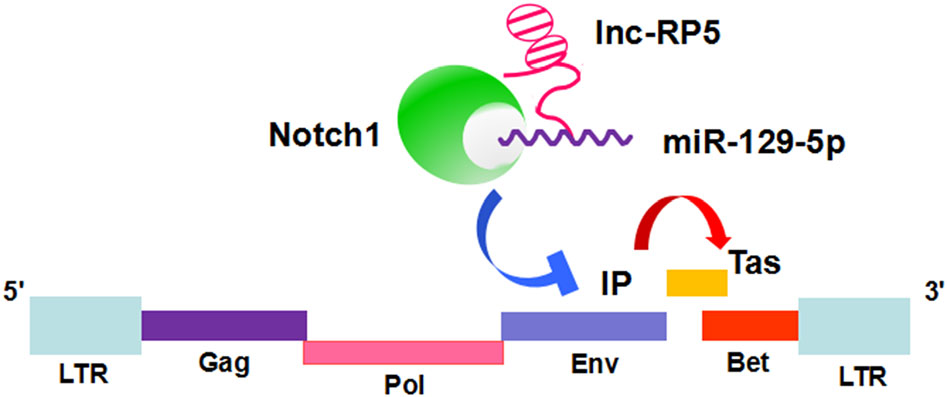
Shanshan Xu, Liujun Chen, Yinglian Tang, Peipei Yuan, Jun Yan, Yingcheng Zheng, Li Huang, Zhi Li, Yan Sun, Song Han, Jun Yin, Qin Pan, Biwen Peng, Xiaohua He and Wanhong Liu. Lnc-RP5 Regulates the miR-129-5p/Notch1/PFV Internal Promoter Axis to Promote the Expression of the Prototype Foamy Virus Transactivator Tas[J]. Virologica Sinica, 2020, 35(1): 73-82. doi: 10.1007/s12250-019-00168-3.
Prototype foamy virus (PFV) is a unique retrovirus that infects animals and humans and does not cause clinical symptoms. Long noncoding RNAs (lncRNAs) are believed to exert multiple regulatory functions during viral infections. Previously, we utilized RNA sequencing (RNA-seq) to characterize and identify the lncRNA lnc-RP5- 1086D14.3.1-1:1 (lnc-RP5), which is markedly decreased in PFV-infected cells. However, little is known about the function of lnc-RP5 during PFV infection. In this study, we identified lnc-RP5 as a regulator of the PFV transcriptional transactivator (Tas). Lnc-RP5 enhanced the activity of the PFV internal promoter (IP). The expression of PFV Tas was found to be promoted by lnc-RP5. Moreover, miR-129-5p was found to be involved in the lnc-RP5-mediated promotion of PFV IP activity, while the Notch1 protein suppressed the activity of PFV IP and the expression of Tas. Our results demonstrate that lnc-RP5 promotes the expression of PFV Tas through the miR-129-5p/Notch1/PFV IP axis. This work provides evidence that host lncRNAs can manipulate PFV replication by employing miRNAs and proteins during an early viral infection.
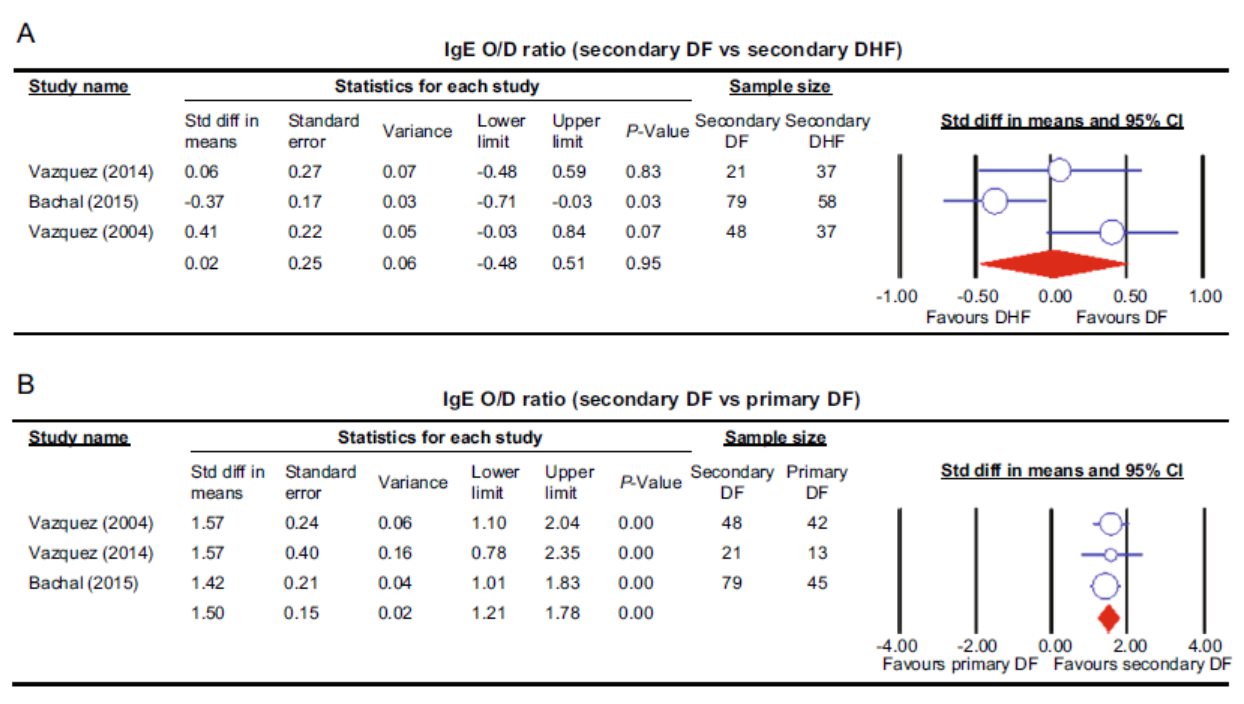
Nguyen Dang Kien, Amr Ehab El-Qushayri, Ali Mahmoud Ahmed, Adnan Safi, Sarah Abdel Mageed, Samar Muhammed Mehyar, Mohammad Rashidul Hashan, Sedighe Karimzadeh, Kenji Hirayama and Nguyen Tien Huy. Association of Allergic Symptoms with Dengue Infection and Severity: A Systematic Review and Meta-analysis[J]. Virologica Sinica, 2020, 35(1): 83-92. doi: 10.1007/s12250-019-00165-6.
The relationship between the severity of dengue infection and allergy is still obscure. We conducted an electronic search across 12 databases for relevant articles reporting allergic symptoms, dengue infection, and dengue classification. These studies were categorized according to dengue severity and allergy symptoms, and a meta-analysis was performed by pooling the studies in each category. Among the included 57 articles, pruritus was the most common allergic sign followed by non-specified allergy and asthma (28.6%, 13%, and 6.5%, respectively). Despite the reported significant association of dengue with pruritus and total IgE level (P < 0.05), in comparison with non-dengue cases and healthy controls, there was no association between the different severe dengue group with pruritus, skin allergy, food allergy or asthma. However, removing the largest study revealed a significant association between asthma with dengue hemorrhagic fever (DHF) rather than dengue fever (DF). In comparison with DF, DHF was associated with IgE positivity. Furthermore, specific-IgE level was higher in secondary DF rather than primary DF. There was a possible association between allergy symptoms and dengue severity progression. Further studies are needed to clarify this association.
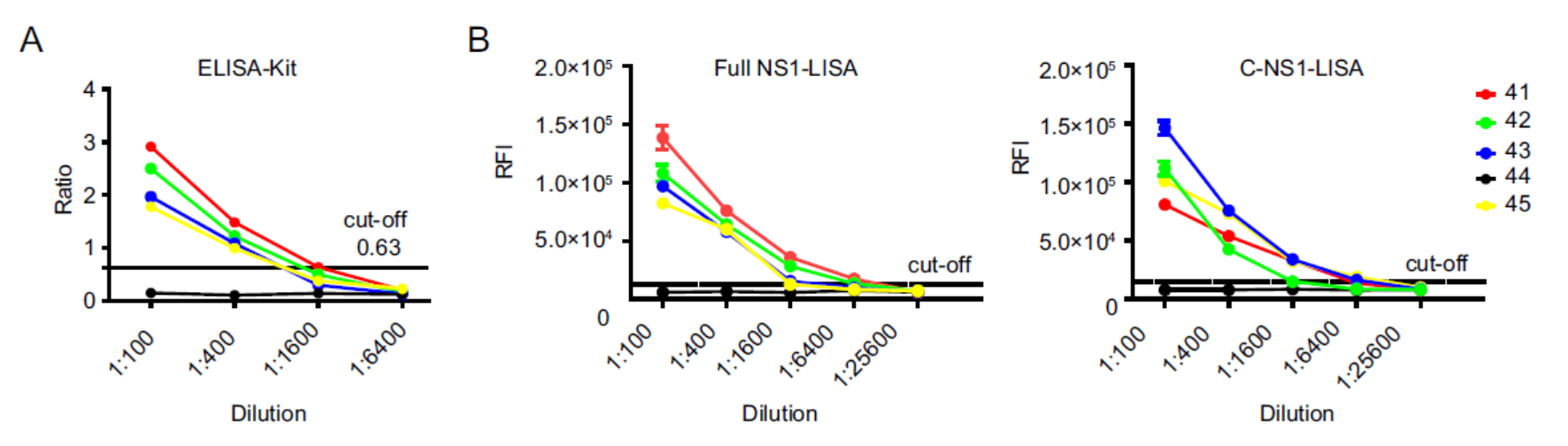
Tianyu Wang, Ying Zhan, De Wu, Zhihai Chen, Wei Wu, Yao Deng, Wenling Wang, Wenjie Tan and Shixing Tang. Development and Evaluation of a Universal and Supersensitive NS1-Based Luciferase Immunosorbent Assay to Detect Zika Virus-Specific IgG[J]. Virologica Sinica, 2020, 35(1): 93-102. doi: 10.1007/s12250-019-00160-x.
Zika virus (ZIKV) causes rash, moderate fever, conjunctivitis, and arthralgia, and has serious connection with neurological complications; therefore, it is a major threat to public health. A rapid and supersensitive method for detecting anti-ZIKV antibodies in humans and animals is thus urgently required. Here, we report an NS1-based luciferase immunosorbent assay (LISA), developed to detect ZIKV-specific IgG. Fusion proteins including a reporter Nano-luciferase (NLuc) and various fragments of ZIKV NS1 protein were expressed in 293 T cells. LISA was performed using the above cell lysates containing the expressed fusion proteins. Sample panels of humans and animals infected with ZIKV were examined for sensitivity of LISA, relative to those of ZIKV RT-PCR, commercial NS1-based ELISA, and micro-neutralization (MN) assays. Specificity and potential cross-reactivity were also evaluated using various convalescent serum samples derived from patients infected with dengue virus (DENV), Japanese encephalitis virus (JEV), and hepatitis C virus (HCV). Results indicated the optimal antigenic domain for anti-ZIKV IgG detection was located within 172–352 amino acids (aa) of ZIKV NS1 protein. NS1-based LISA performs better than commercial ELISA in anti-ZIKV IgG detection. LISA was shown to be at least fourfold more sensitive than commercial ELISA, and could detect anti-ZIKV IgG in various animal hosts without the need of species-specific labeled antibody. This novel assay is potentially useful for the rapid and sensitive detection of anti-ZIKV IgG in human and animal samples.
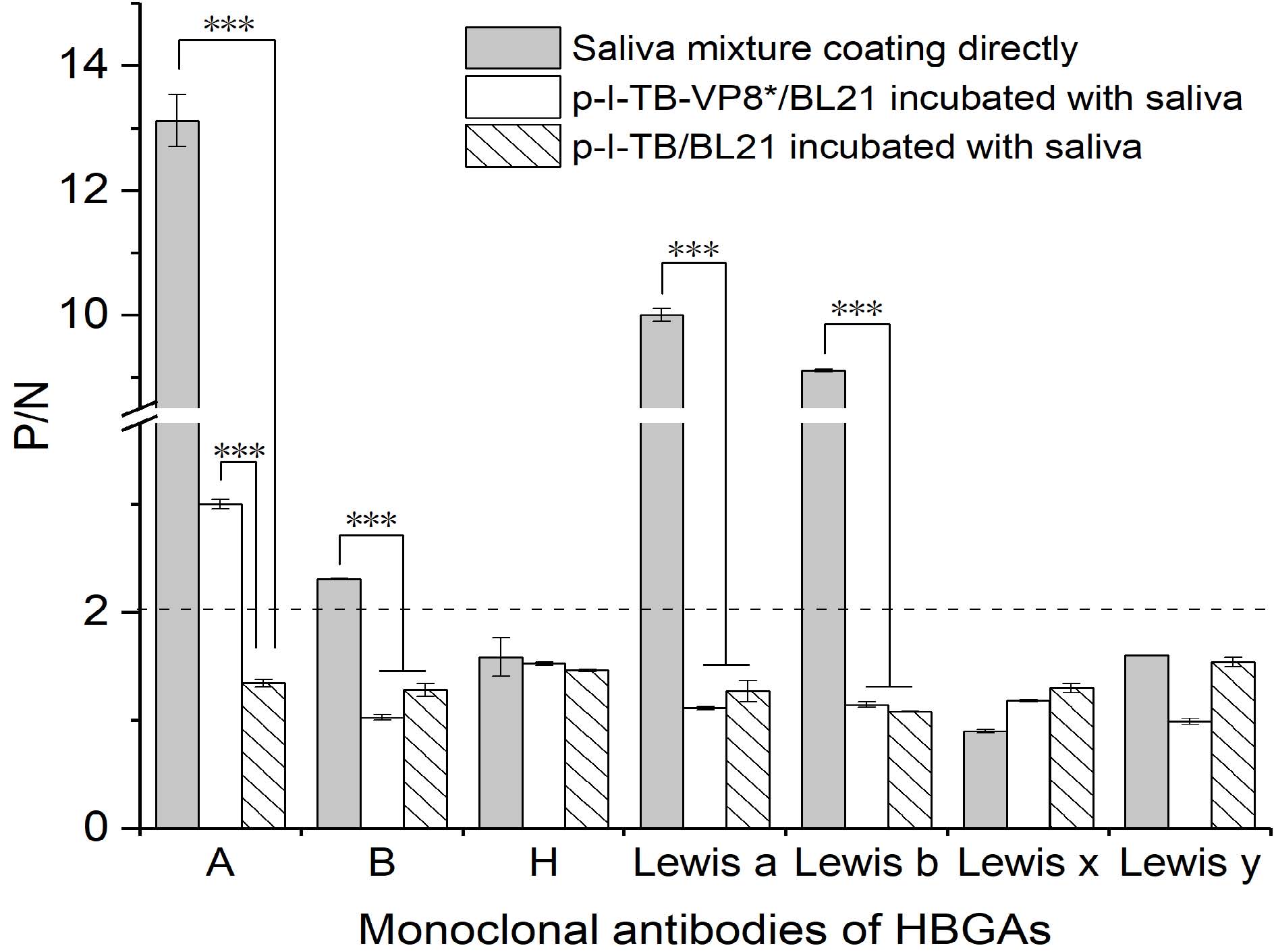
Danlei Liu, Haoran Geng, Zilei Zhang, Yifan Xing, Danlu Yang, Zhicheng Liu and Dapeng Wang. An Effective Platform for Exploring Rotavirus Receptors by Bacterial Surface Display System[J]. Virologica Sinica, 2020, 35(1): 103-109. doi: 10.1007/s12250-019-00174-5.
Rotavirus (RV) is a major foodborne pathogen. For RV prevention and control, it is a key to uncover the interaction mechanism between virus and its receptors. However, it is hard to specially purify the viral receptors, including histo-blood group antigens (HBGAs). Previously, the protruding domain protein (P protein) of human norovirus (genotype II.4) was displayed on the surface of Escherichia coli, and it specifically recognized and captured the viral ligands. In order to further verify the feasibility of the system, P protein was replaced by VP8* of RV (G9P[8]) in this study. In the system, VP8* could be correctly released by thrombin treatment with antigenicity retaining, which was confirmed by Western blot and Enzyme-Linked Immunosorbent Assays. Type A HBGAs from porcine gastric mucin (PGM) were recognized and captured by this system. From saliva mixture, the captured viral receptor bound with displayed VP8* was confirmed positive with monoclonal antibody against type A HBGAs. It indicated that the target ligands could be easily separated from the complex matrix. These results demonstrate that the bacterial surface display system will be an effective platform to explore viral receptors/ligands from cell lines or food matrix.
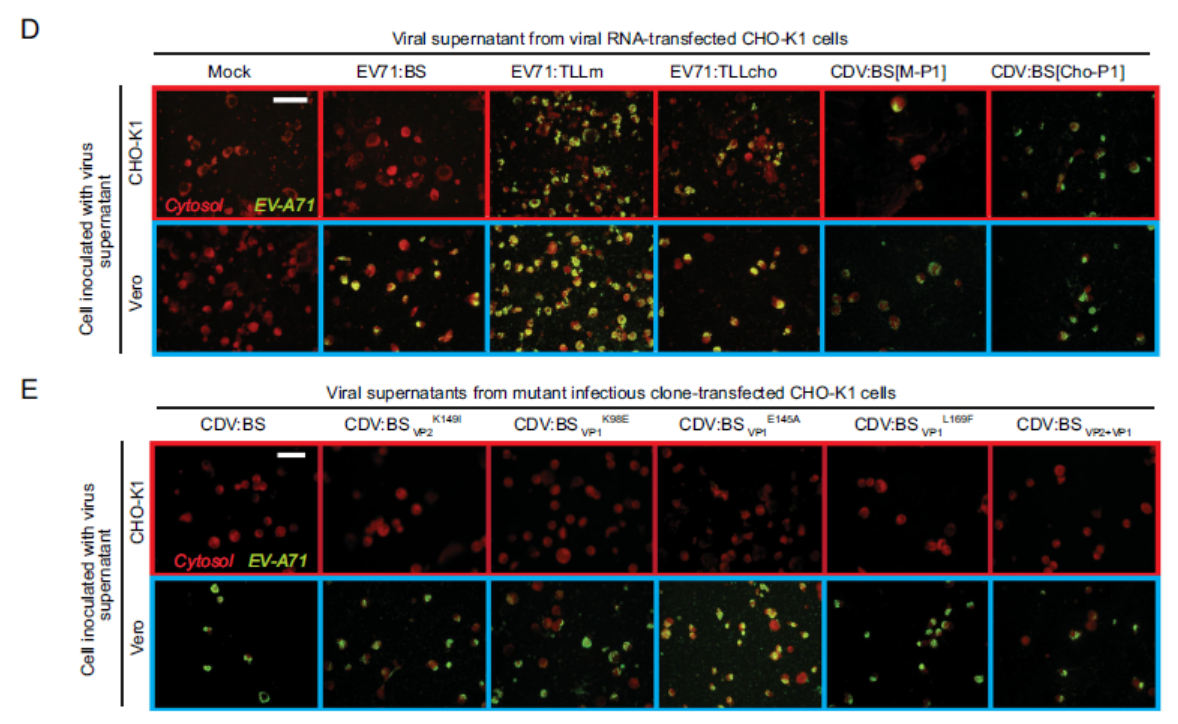
Carla Bianca Luena Victorio, Yishi Xu, Qimei Ng, Vincent T. K. Chow and Kaw Bing Chua. Reverse Genetic Analysis of Adaptive Mutations within the Capsid Proteins of Enterovirus 71 (EV-A71) Strains Necessary for Infection of CHO-K1 Cells[J]. Virologica Sinica, 2020, 35(1): 110-114. doi: 10.1007/s12250-019-00167-4.
Enterovirus 71 (EV-A71) is a human pathogen that does not naturally infect rodent cells. However, virus strains that productively infect rodent cells could be produced by serial passage in a rodent cell line. The resulting viruses are useful tools to identify molecular determinants of adaptation to a new host species. We adapted an EV-A71 clinical isolate (EV71:BS) in Chinese hamster ovary (CHO-K1) cells and sequenced the genome of the resulting virus. We explored the contribution of viral capsid mutations that may have contributed to the CHO-adapted phenotype by introducing the mutations surrounding the receptor-binding canyon region into the genome of EV71:BS by reverse genetics tools. We subsequently assessed whether the resulting mutant viruses productively infected CHO-K1 cells. Contrary to previous reports in the literature, our findings demonstrate that a single amino acid substitution in capsid VP2149 K→I is not sufficient for the resulting mutant virus to infect CHO-K1 cells. Moreover, a mutant virus harbouring other capsid mutations we evaluated (VP1 K98→E, E145→A, and L169→F) also did not infect CHO-K1 cells. These findings strongly suggest that cooperation of various adaptive mutations in the capsid is necessary to enable successful EV-A71 infection of a different host species.
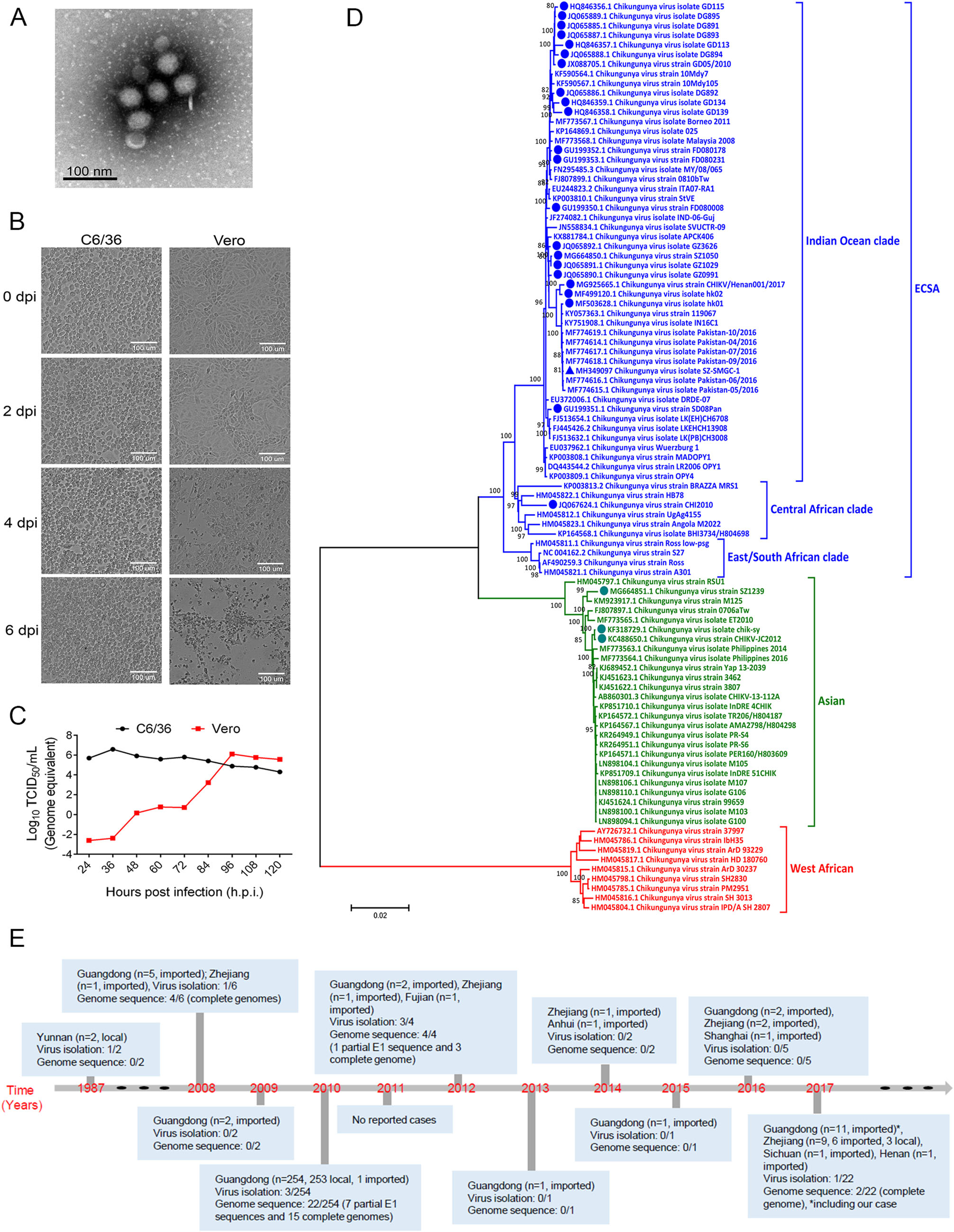
Yang Yang, Zhixiang Xu, Haixia Zheng, Jingdong Song, Ying Wu, Zhou Tong, Jing Yuan, Gary Wong, William J. Liu, Yuhai Bi, Yingxia Liu and George F. Gao. Genetic and Phylogenetic Characterization of a Chikungunya Virus Imported into Shenzhen, China[J]. Virologica Sinica, 2020, 35(1): 115-119. doi: 10.1007/s12250-019-00166-5.
Chikungunya virus (CHIKV) is a mosquito-borne virus belonging to the family Togaviridae, genus Alphavirus, and was first isolated in Tanzania in the 1950s (Silva and Dermody 2017; Weaver and Lecuit 2015). Human infections with CHIKV typically result in a rapid-onset febrile disease, with symptoms that include fever, headache, rash, severe joint and muscle pain, as well as prolonged periods of disability in some patients (Weaver and Lecuit 2015; Silva and Dermody 2017). Unlike other arboviral diseases such as dengue and Zika fever, the majority of CHIKV infections in humans results in clinical symptoms, with ~15% of human infections who are asymptomatic with seroconversion (Schwartz and Albert 2010; Weaver and Lecuit 2015). CHIKV has been reported in more than 110 countries and territories in Asia, Africa, Europe, and the Americas (CDC 2019), and has evolved into three main lineages, including West African, Asian, and East Central South African (ECSA) (Silva and Dermody 2017; Weaver and Lecuit 2015). The ECSA lineage emerged in Kenya during 2004 and spread to islands in the Indian Ocean, resulting in a large epidemic (Silva and Dermody 2017; Weaver and Lecuit 2015). The first report of CHIKV in China was in 1987 in Yunnan Province (Wu et al. 2012). Since then, several sporadic cases of imported CHIKV infections and a local outbreak have been documented in Guangdong Province (Qiaoli et al. 2012; Wu et al. 2012; Sun et al. 2013; Wu et al. 2013). Here, we describe and present the phylogenetic as well as genetic analysis of a novel CHIKV isolate from a patient with fever and chills traveling from Pakistan to Shenzhen, China in April 2017.
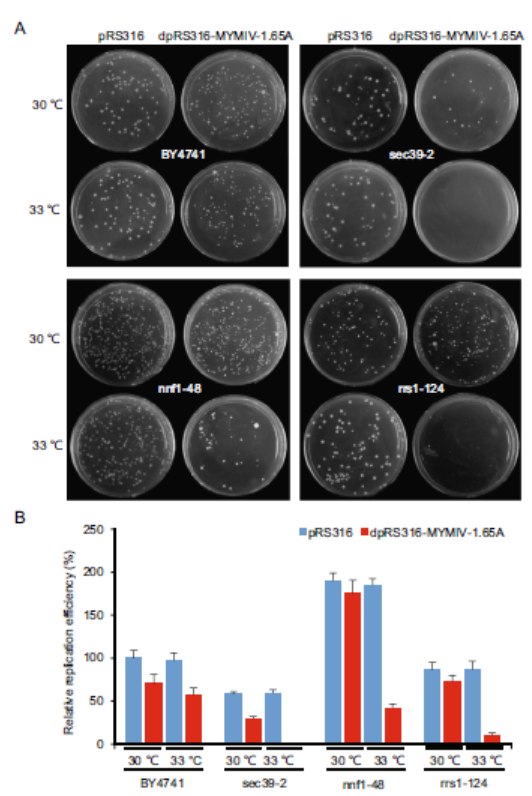
Fangfang Li, Xiongbiao Xu, Zhenghe Li, Yaqin Wang and Xueping Zhou. Identification of Yeast Factors Involved in the Replication of Mungbean Yellow Mosaic India Virus Using Yeast Temperature-Sensitive Mutants[J]. Virologica Sinica, 2020, 35(1): 120-123. doi: 10.1007/s12250-019-00154-9.
Geminivirus replicates its genomic DNA in host cell nucleus through rolling-circle replication (RCR), which relies heavily on host's DNA replication machinery/factors to fulfill this process. Saccharomyces cerevisiae (yeast) as a model has been used to explore the replication mechanism of many viruses, and the favorable yeast temperature-sensitive (ts) mutant library of essential genes has helped to identify many important host factors participating in positive RNA virus replication. Here, we identified 131 host proteins whose mutations had important roles in the replication of Mungbean yellow mosaic India virus (MYMIV) in yeast cells using these yeast ts mutants. The identified proteins could be classified into 20 distinct categories based on their known cellular/biochemical functions, among which 13 proteins (10%), 14 proteins (11%) and 16 proteins (12%) are involved in DNA replication, cell division cycle regulation, and protein transport and secretion, respectively. In addition, the identified 39 essential yeast genes which have been reported previously to affect the replication of Tomato bushy stunt virus were also involved in MYMIV replication, indicating the universal feasibility by screening this ts mutant library. Given the importance of DNA replication in viral infection process, our data provided a high throughput method to identify host factors for geminivirus replication using ts mutant library and we identified many host factors affecting MYMIV replication, which could serve as candidate genes for anti-viral plant breeding.
Nguyen Dang Kien, Amr Ehab El-Qushayri, Ali Mahmoud Ahmed, Adnan Safi, Sarah Abdel Mageed, Samar Muhammed Mehyar, Mohammad Rashidul Hashan, Sedighe Karimzadeh, Kenji Hirayama and Nguyen Tien Huy. Correction to: Association of Allergic Symptoms with Dengue Infection and Severity: A Systematic Review and Meta-analysis[J]. Virologica Sinica, 2020, 35(1): 124-124. doi: 10.1007/s12250-019-00180-7.
The spelling of the fifth author's name was misspelled. The byline should appear as shown above. The original article has been corrected.