














HTML
-
Two predominant circulating HIV-1 recombinants, CRF07_BC and CRF08_BC, which are the recombinants of clade B' and C, have been proven to account for 55.6% of all HIV-1 infections in China (He et al. 2012). Though their genomes mainly come from subtype C, portions of LTR, gag, pol, env, nef and tat are derived from subtype B (Thai-B) (Su et al. 2000; Yang et al. 2003). Previous research suggested that CRF07/08_BC isolates had lower in vitro replication capacities compared with clade B isolates (Ma et al. 2009), and that the CRF07_BC infected individuals often showed lower viral loads as well as slower disease progressions compared with the clade B infected individuals (Huang et al. 2014). However, CRF07_BC has still become a major circulating HIV-1 strain in China. Thus, the investigation of the unique genetic characteristics that potentially impact replication capacity helps to elaborate the wide transmission of clade BC isolates.
HIV-1 Rev is a 19 kDa phosphoprotein primarily localized in the nucleolus/nucleus. At the later stage of the expression of HIV-1 mRNA, Rev binds to Rev-responseelement (RRE), a 351-nucleotide structured region in the unspliced viral mRNAs and form the Rev-RRE ribonucleoprotein (RNP) (Pollard and Malim 1998). After that, a host nuclear export protein, CRM1, is recruited by RNP via a nuclear export sequence (NES) within Rev in order to facilitate the nuclear exportation, stabilization, and translation of the viral mRNAs (Booth et al. 2014). In this way, Rev quickly shuttles between the nucleus and cytoplasm (Li et al. 2005).
Naturally occurring sequence variations within Rev have been reported to influence Rev function and viral replication (Jackson et al. 2016). Rev variants derived from HIV-1 infected long term non-survivors showed lower functional capacities compared with the laboratory-adapted strains (Iversen et al. 1995; Churchill et al. 2007). Mutations that were not present in the known functional domains of Rev also exerted an effect on the function of Rev (Phuphuakrat and Auewarakul 2005; Phuphuakrat et al. 2005). A seven amino-acid deletion of p6 gag has been identified in clade BC involved in the virus replication (Zhefeng et al. 2011; Huang et al. 2014; Wu et al. 2017). In our previous study, we observed that a premature stop codon at residue 101 causes a truncation of 16 AAs in Rev of clade BC (Wang et al. 2013). Thus, we hypothesis that the unique truncation of Rev of clade BC may be involved in Rev function as well as viral replication capacity. However, no studies were conducted on the effect of this truncation on Rev function and viral replication. In this study, we investigated the effects of a stop codon at residue 101 on Rev activity and viral replication in HIV-1 clade BC and B strains.
-
All available HIV-1 infected individual-derived Rev or Rev exon2 sequences for clade A, B, C, D, AE, G, AG and BC were obtained from the Los Alamos HIV Sequence Database (http://www.hiv.lanl.gov), and one sequence was chosen per infected individual. The number of selected sequences of each clade is shown in Table 1. BioEdit v7.0.9 was used to adjust alignment and to produce consensus sequence for each clade.
Clade Number of sequences presenting allele variance Total number Stop codon Q E S K R P H V I CRF07_BC 140 135 1 4 0 0 0 0 0 0 0 CRF08_BC 65 62 2 1 0 0 0 0 0 0 0 CRF57_BC 7 7 0 0 0 0 0 0 0 0 0 CRF60_BC 5 3 2 0 0 0 0 0 0 0 0 CRF61_BC 3 3 0 0 0 0 0 0 0 0 0 CRF62_BC 3 3 0 0 0 0 0 0 0 0 0 CRF64_BC 8 8 0 0 0 0 0 0 0 0 0 C(Asia) 182 160 13 9 0 0 0 0 0 0 0 C(Africa) 1306 1217 37 45 2 1 1 1 0 1 1 B 7023 8 6780 198 0 4 0 31 2 0 0 CRF01_AE 961 9 941 7 0 0 1 2 1 0 0 D 194 1 192 0 0 1 0 0 0 0 0 CRF02_AG 271 16 251 2 0 0 0 2 0 0 0 A 18 8 9 0 1 0 0 0 0 0 0 G 123 7 111 3 0 1 0 0 1 0 0 Table 1. Number of Rev sequences presenting allele variance at residue 101 in the different clades.
-
Rev gene of CRF07_BC isolate (pXJDC13), carried the stop codon at the end of C-terminal, was cloned into pcDNA3.1-TOPO-V5-His vector (Invitrogen, Catalog: K4900-01, USA) to generate clade BC wild type Rev clone (BC WT Rev). The full length Rev gene in which the stop codon at residue 101 was replaced by Q, was chemically synthesized and named BC Rev Stop101Q. Rev mutant of Clade B (pSV-Rev Q101Stop) was generated based on the backbone of BH10 isolate Rev expression vector pSV-Rev (Smith et al. 1990).
Rev genes of pXJDC13 and BH10 and their mutations (without stop codon at the end of C-terminal) were separately cloned into pcDNA3.1-TOPO-V5-His vector to generate Rev expression plasmids with His-tag (BC WT Rev-His/BC Rev Stop101Q-His and B WT Rev-His/B Rev Q101Stop-His).
-
Rev expression plasmids with His-tag (5 μg) were transfected into 5 × 105 HEK 293T cells. Fourty-eight hours after transfection, transfected cell samples were homogenized in a lysis buffer containing complete protease inhibitor cocktail (Kangweishiji, Nanjing, China) and then centrifuged at 12, 000g, 4 ℃ for 15 min for supernatant collection. Protein concentrations were determined by the use of the BCA protein assay kit (Kangweishiji, Nanjing, China). Equal amount of total protein (30 μg) were resolved on 10% SDS PAGE gel and transferred to PVDF membranes. Membranes were blocked with 5% skim milk/PBST overnight, then were incubated with anti- 6 × His mouse monoclonal antibody (1:1000 dilution) (Kangweishiji, Nanjing, China) and anti-β-actin mouse monoclonal antibody (1:1000 dilution) (Boaolong, Beijing, China), respectively. Horseradish peroxidase-linked antimouse-IgG (1:10, 000) (Boaolong, Beijing, China) were employed to detect the results with PierceTM ECL Western Blotting Kit (Pierce, USA) and membrane was imaged in a Molecular Imager CheminDocTMXRS + Systems (Biorad, USA). Image was analyzed using Image J-win32 software. The Rev signal was normalized to β-actin levels measured in the same lane of the same Western blot.
-
HEK 293T cells (105/well) were seeded into 24-well tissue culture plates in 1 mL of culture medium. After 17 h, using the Sino-infection transfection reagent (Sino Biological Inc., China), each well was transfected with 1 μg Gag-Pol-RRE plasmid plus 15 ng, 30 ng or 60 ng of Rev expression plasmid (B WT Rev, B Rev Q101Stop, BC WT Rev or BC Rev Stop101Q), and 200 ng CMV-EGFP plasmid (transfection control), respectively. Supernatants were harvested at 48 h post transfection and filtered by 0.45 lm filter for p24 quantification using ELISA. The p24 antigen was normalized to transfection efficiency as determined by percentage of EGFP expressing cells measured using FACSCalibur (Becton–Dickinson, USA).
-
A 1800 bp fragment spanning from V3 to Nef was amplified by high-fidelity PCR from a CRF07_BC infectious clone pXJDC13 (Wang et al. 2013), using pairs of primers carrying restriction enzyme sites (ClaI-F and BamHI-R) (Supplementary Table S1). Then the PCR products were sub-cloned into the pGEM-T vector (Promga, USA), and mutations were introduced subsequently using QuikChange Multi Site-Directed Mutagenesis Kit with two paired primers (BC-E117Stop-F and BC-E117Stop-R combined with BC-Stop101Q-F and BC-Stop101Q-R) (Supplementary Table S1). The mutated fragment was digested by Cla I and BamH I and ligated into plasmid pXJDC13 to generate CRF07_BC mutated clone pXJDC13-Stop101Q.
AT1 and AT2 are two pNL4-3 based clade B infectious clones in which nef was substituted by Thy1.1 (Rat CD90.1) and Thy 1.2 (Mouse CD90.2) (Dykes et al. 2006) separately. A subclone carrying Rev from clone AT1 was initially constructed using the primers, B-BmtI-F and B-HpaI-R (Supplementary Table S1). The mutations were introduced using Quicession plasmids with Hisk-change Multi Site-Directed Mutagenesis Kit (Stratagene, USA) with primers (B-BH10- Q101Stop-F and B-BH10-Q101Stop-R). Following double digestion of Bmt I and Hpa I enzymes, mutated Rev was ligated into the AT1 to generate clade B mutated clone AT1-Q101Stop.
HEK 293T cells were maintained in the DMEM containing 10% FCS, 100 mg/mL of streptomycin, 100 IU/mL of penicillin, full-length genome plasmid (5 μg) was transfected into 5 × 106 HEK 293T cells using the Sinoinfection transfection reagent (Sino Biological Inc., China) in a six-well plate to generate clone-derived viruses. The supernatant was collected 48 h post infection, filtered through a 0.2 lm filter, and stored at - 80 ℃ for p24 quantification.
-
Viruses pXJDC13 and pXJDC13-Stop101Q were propogated in PBMCs for they were CCR5-tropic primary isolates (Wang et al. 2013). PBMCs were isolated from the healthy HIV-1 seronegative donors using the Ficoll-Hypaque gradient method. The p24 antigen quantified virus (10 ng) was used to infect 3 × 106 phytohemaglutinin (PHA)-stimulated PBMCs in triplicate for 2 h at 37 ℃ in 1 mL total volume, then the supernatant was removed and cells were washed twice by 10 mL phosphate buffer saline (PBS). The cells were then cultured in 3 mL of complete RPMI 1640 medium (containing 10% FCS and 10 IU/mL IL-2) in a 25 cm2 flask (Corning, USA). Supernatant (1 mL) was substituted by fresh medium every 3–4 days, and 3 × 106 PHA-stimulated fresh PBMCs were seeded into the flasks every 7 days, and the supernatant obtained was passed through a 0.2 lm filter and stored at - 80 ℃.
Viruses AT1 and AT1-Q101Stop were replicated in a human T cell type, PM1 cells. The p24 antigen quantified virus (10 ng) was inoculated into 7 × 105 PM1 cells in 1 mL total volume for 2 h at 37 ℃, after which cells were washed with 5 mL PBS twice. Then the cells were grown in 2 mL of complete RPMI 1640 medium, and 0.5 mL medium was replaced by fresh medium at days 3, 5 and 7 post-infection for p24 quantification.
-
The growth competition assay was conducted as previously described (Dykes et al. 2006; Wang et al. 2017). Cells were harvested by centrifuging on days 3, 4, 5 and 6 post-infection. For each sample, cells (5 × 105) were stained with anti-Thy1.1-FITC antibody (BD Biosciences, USA) plus 7AAD (BD Biosciences, USA), anti-Thy1.2-PE antibody (BD Biosciences, USA) plus 7AAD, and antiThy1.1 antibody combined with anti-Thy1.2 antibody plus 7AAD, respectively. The anti-CD4 antibody-PE plus 7AAD, anti-CD4 antibody-FITC plus 7AAD, and 7AAD stained cells were set as controls, separately. Following the antibodies incubation in the dark for 30 min on ice, stained cells were washed with PBS and fixed with 4% paraformaldehyde in PBS for flow cytometry performed as described (Dykes et al. 2006; Wang et al. 2017). The data was analyzed using CellQuest software (Becton–Dickinson, San Jose, CA) and FlowJo software (Treestar, USA). The percentages of cells infected with virus mutant, wild-type virus and both viruses are labeled in the lower right, upper left and upper right quadrants, respectively.
-
Relative fitness of virus was calculated using the formula: Fitness: 2-Point Calculation in Competitive Virus Fitness (http://bis.urmc.rochester.edu/TFitness/FitnessTwo.aspx) (Ma et al. 2010) using the formula:
$1 + {\rm{s}} = {\rm{exp}}\left({\frac{1}{3} \times {\rm{ln}}\frac{{{\rm{Tm}}({\rm{t}}6){\rm{Tw}}({\rm{t}}3)}}{{{\rm{Tw}}({\rm{t}}6){\rm{Tm}}({\rm{t}}3)}}} \right)$ -
Student's t test was used to assess the statistical difference between two groups using Graphpad Prism (version 5.0, GraphPad, Inc.). Two-way ANOVA was applied to determine the statistical significance of replication curves of virus, set as P < 0.05 (two tailed value). Results are shown as the mean ± SD of independently triplicate experiments.
Rev Sequence Analysis for Each HIV-1 Clade
Rev Gene Cloning, Plasmid Construction and Mutagenesis
Western Blotting
Measurement of Rev-RRE Activity
Virus Production
Replication Capacities of Virus Constructs
Growth Competition Assay and Flow Cytometry Analysis
Calculation of Viral Relative Fitness
Statistical Analysis
-
Briefly, 1488 sequences of clade C prevalent in Asia and Africa, 7023 sequences of clade B, 231 sequences of clade BC isolates, 961 sequences of clade AE and 606 sequences of other clades (A, D, CRF02_AG and G) were collected (Table 1). Sequences of each clade were aligned and consensus Rev sequences of each clade were produced respectively. Considering the CRF07_BC and CRF08_BC were products of recombination of clade B' and clade C virus prevalent in Asia, we divided clade C virus into two sub-groups: C strains isolated in Asia (C-Asia) or in Africa (C-Africa). As shown in Fig. 1A, the key domains, e.g., the first oligomerization domain (OD1), arginine rich motif (ARM), the second oligomerization domain (OD2) and nuclear export signal (NES) are highly conservative in clade BC and clade C. For clade B and clade D, a deletion of 7 AAs (QSQGVET) in the C-terminal is observed. For clade C and clade BC, a premature stop codon within Rev has been shown to account for a truncation of 16 AAs.
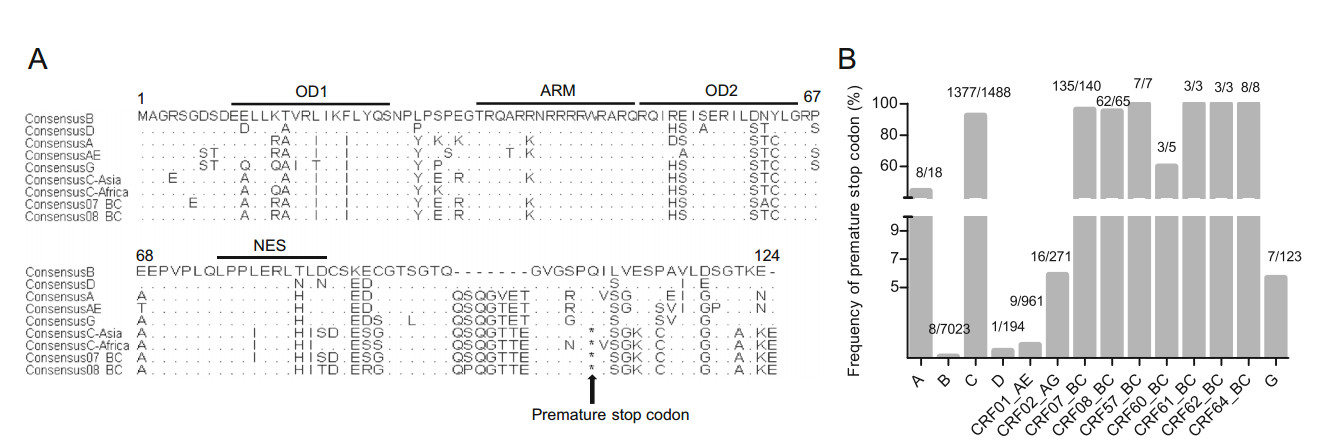
Figure 1. A Alignment of Rev amino acid sequences of different HIV-1 clades. Key domains of Rev are also marked by lines. Substitutions or deletions within each region are shown; conserved amino acids are indicated with point (.), deletions are shown with dash (-) and the star (*) represents the premature stop codon. Clade D have the same deletions (QSQGVET) as clade B. B Frequency of premature stop codon within Rev of different HIV-1 clades.
To investigate the frequency of this premature stop codon in the different clades, allele variations at residue 101 were identified and frequencies of the premature stop codon were then calculated, respectively. Interestingly, a premature stop codon is shown to exist in each clade (Table 1). A premature stop codon is present at high frequencies in CRF07_BC and CRF08_BC isolates, i.e., 96.40% (135/140) and 95.38% (62/65) (Fig. 1B). Similar to the CRF07_BC and CRF08_BC, 94.27% (1377/1488) of clade C isolates carry premature stop codons within Rev. In contrast, clade B isolates have the lowest frequency (0.11%, 8/7023) of a premature stop codon (Fig. 1B).
-
To discover whether the premature stop codon influences Rev expression level, Rev expression vector of clade BC and its mutant were constructed with pcDNA3.1-TOPO-V5-His vector (Fig. 2A). HEK 293T cells were transfected with these two expression plasmids separately, and collected to determine Rev expression levels by Western blotting at 48 h post transfection (Fig. 2B). We found that Rev with Q101 mutation did not show higher expression level than wild-type Rev with a statistical significant difference (Fig. 2C).
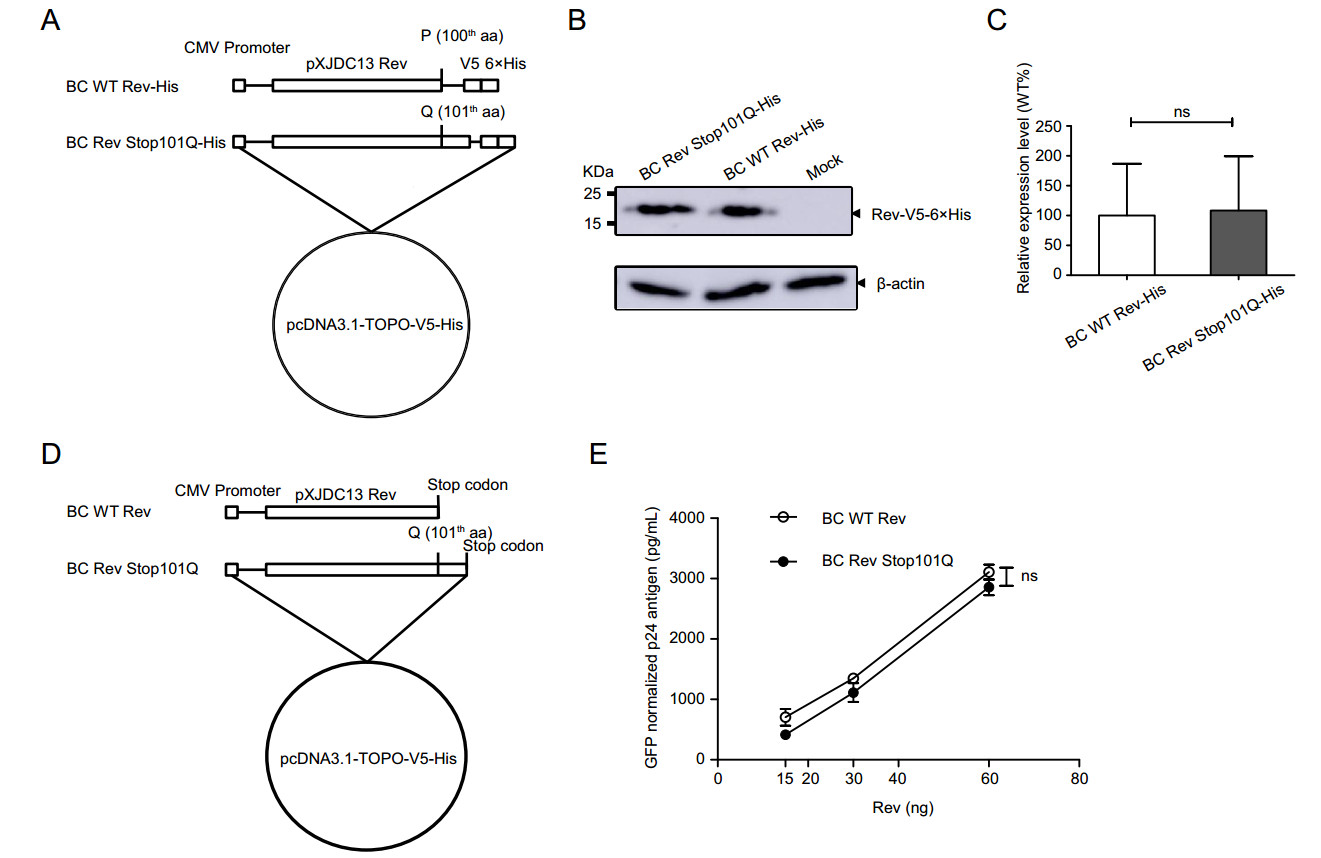
Figure 2. Construction of CRF07_BC Rev expression plasmids and evaluation of their expression levels and Rev-RRE activities. A BC WT Rev-His: clade BC wild type Rev gene sequence; BC Rev Stop101Q-His: full length Rev gene in which the stop codon at residue 101 was replaced by Q. These two inserts were cloned into pcDNA3.1-TOPO-V5-His vector in frame with V5-His tag, separately. B Analysis of expression levels of Rev constructs with His Tag using Western blotting. C Relative expression levels of Rev mutant to wild type. Each bar represents the average and standard deviation of at least three replicate experiments. D BC WT Rev: clade BC wild type Rev gene sequence carrying a premature stop codon; BC Rev Stop101Q: full length Rev gene in which the premature stop codon at residue 101 was replaced by Q, and a TAG stop codon was contained at the end of sequence. These two fragments were separately cloned into pcDNA3.1-TOPO-V5-His vector to produce proteins without V5-His tag. E Measurement of Rev-RRE activity of WT and mutated CRF07_BC Rev protein. HEK 293T cells were transfected with clade BC Gag-Pol-RRE plasmid plus Rev expression plasmid (BC WT Rev or BC Rev Stop101Q). CMV-EGFP plasmid was co-transfected as control. Supernatants were harvested 48 h post transfection for p24 quantification using ELISA. The p24 antigen was normalized to transfection efficiency as determined by percentage of EGFP expressing cells measured using flow cytometry. Data is representative of at least three independent experiments. ns: no significance.
In order to explore the effect of the premature stop codon on clade BC Rev-RRE activity, BC Rev expression vector (BC WT Rev or BC Rev Stop101Q) (Fig. 2D) was co-transfected with BC Gag-Pol-RRE expression plasmid and CMV-EGFP plasmid into HEK 293T cells. Supernatant was harvested for p24 antigen quantification at 48 h post transfection. Cells were collected for flow cytometry to determine GFP-positive rate. P24 antigen was normalized to percentage of GFP-positive cells. It is noted that mutated Rev has not displayed higher biological activities compared with wild-type Rev (Fig. 2E). These data thus together indicated that premature stop codon had no effect on expression level of Rev and Rev-RRE activity, for clade BC.
-
For Q at position 101 within Rev is present in strains from each HIV-1 clade, we speculated that substitution of stop codon by Q may affect the replication activity of clade BC. To determine whether the substitution of premature stop codon alters viral replication capacity, virus carrying Q101 in Rev (pXJDC13 Stop101Q) was constructed (Fig. 3A). The p24 antigen normalized virus pXJDC13 and pXJDC13 Stop101Q (10 ng p24 each) were used to infect PHA-stimulated PBMCs independently to reveal the effects of mutations at residue 101 on the viral replication capacity. P24 antigen was quantified from the supernatant at the different time points post infection, and replication curves of two viruses were mapped (Fig. 3B). As shown in Fig. 3B, no statistically significance on the replication curve was observed between mutated and wild-type viruses. The above results suggested that premature stop codon within Rev had no effect on Rev expression, Rev-RRE activity and viral replication consequently.
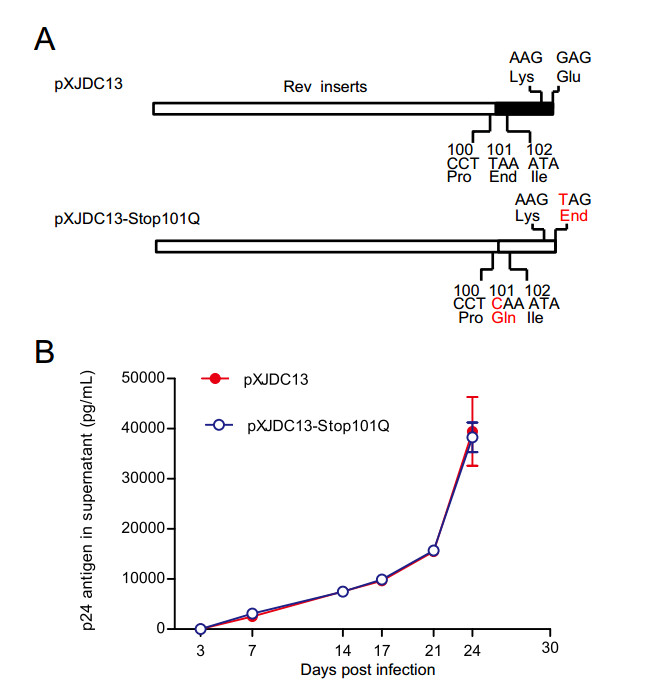
Figure 3. Replication capacities of CRF07_BC wild type and mutated virus in PBMCs. A Diagrams of methods to construct mutant of CRF07_BC (pXJDC13-Stop101Q). The red character indicates the mutated base or amino acid. B Replication capacities of virus in PBMCs. CRF07_BC wild type (pXJDC13) and mutated virus (pXJDC13-Stop101Q) (10 ng p24 antigen) were used to infect 3 × 106 phytohemaglutinin (PHA)-stimulated PBMCs in triplicate. The cells were then cultured in 3 mL of complete RPMI 1640 medium (containing 10% FCS and 10 IU/mL IL-2) in a 25 cm2 flask (Corning, USA). Supernatant (1 mL) was substituted for p24 antigen quantification at days 3, 7, 14, 17, 21 and 24 post infection, and 3 × 106 PHA-stimulated fresh PBMCs were seeded into the flasks every 7 days. Data is representative of at least three independent experiments.
-
To discover whether the stop codon at residue 101 of clade B Rev influences Rev expression and its function, Q101 in Rev of BH10 isolate was mutated to a stop codon (Fig. 4A). Forty-eight hours after transfection, the expression levels of B WT Rev-His and B Rev Q101Stop-His were detected by Western blotting (Fig. 4B). Compared to the wild-type, Rev expression level of mutant decreased by over 90% with a statistical significance (Fig. 4C).
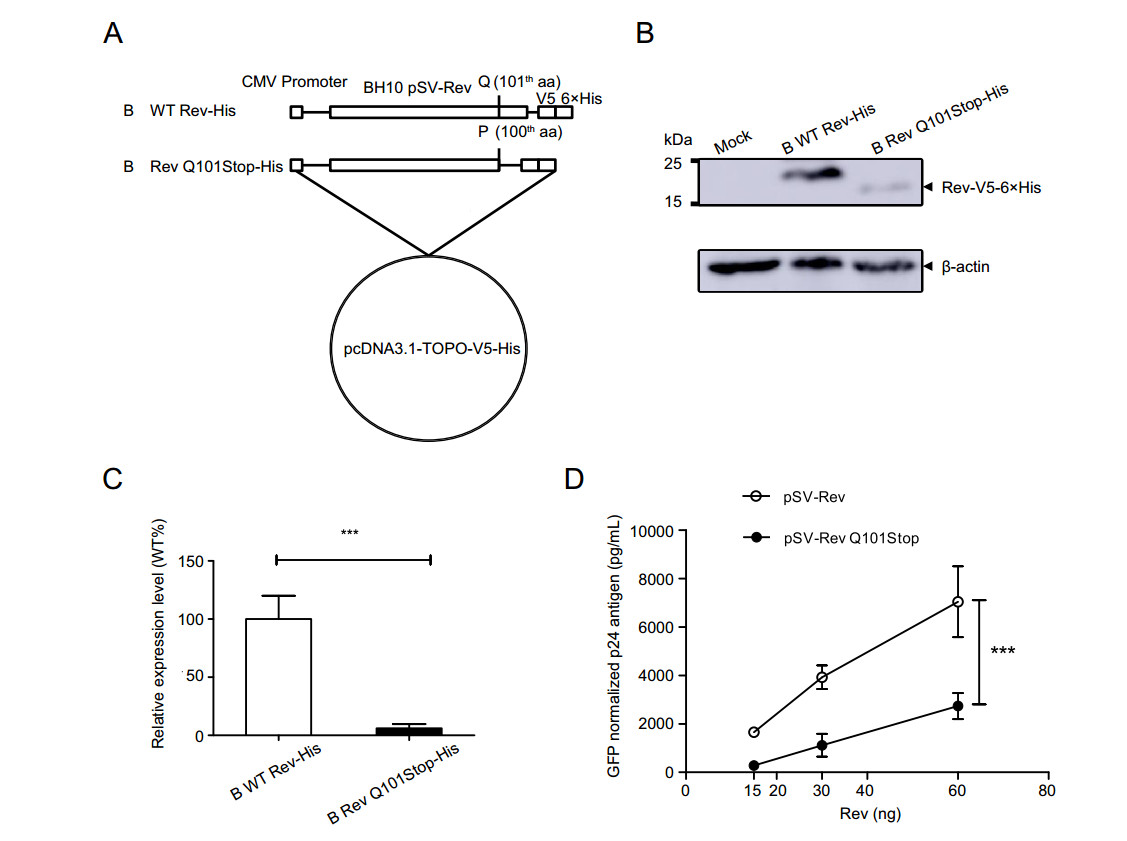
Figure 4. Construction of clade B Rev expression plasmids and evaluation their expression levels and Rev-RRE activities. A B WT Rev-His: full length clade B wild type Rev gene sequence; B Rev Q101Stop-His, a shorter Rev gene without C-terminal after 100-th aa. These two inserts were respectively cloned into pcDNA3.1-TOPO-V5-His vector to generate Rev proteins with V5-His tag. B Analysis of expression levels of clade B Rev constructs with His Tag using Western blotting. C Relative expression levels of clade B Rev mutant to wild type. Each bar represents the average and standard deviation of at least three replicate experiments. ***P < 0.001. D Measurement of Rev-RRE activity of WT and mutated Clade B Rev protein. HEK 293T cells were transfected with clade B Gag-Pol-RRE plasmid plus Rev expression plasmid (pSV-Rev and pSV-Rev Q101Stop). CMV-EGFP plasmid was co-transfected as control. Supernatants were harvested 48 h post transfection for p24 quantification using ELISA. The p24 antigen was normalized to transfection efficiency as determined by percentage of EGFP expressing cells measured using flow cytometry. Data is representative of at least three independent experiments. ***P < 0.001.
Then, HEK 293T cells were co-transfected with CMV-EGFP plasmid, clade B Gag-Pol-RRE plasmid and Rev expression plasmid (B WT Rev or B Rev Q101Stop) to investigate the effect of stop codon on clade B Rev-RRE activity. Result showed that the differences of normalized p24 antigen concentrations between the wild type and mutant were significant at doses of 15, 30 and 60 ng (Fig. 4D). It suggested that Q101Stop within clade B Rev significantly suppressed Rev-RRE activity.
-
To study the effect of a stop codon at residue 101 within clade B Rev on viral replication, virus carrying the mutation of Q101Stop was constructed (Fig. 5A). The same amount of p24 antigen HIV virus (AT1 and AT1-Q101Stop) was used to infect PM1 cells separately. At different time points post infection, p24 antigen in supernatant was measured. Virus with Q101Stop was almost deprived of the replication capacity, and wild-type virus exhibited the highest replication capacity with a statistical significance (Fig. 5B).
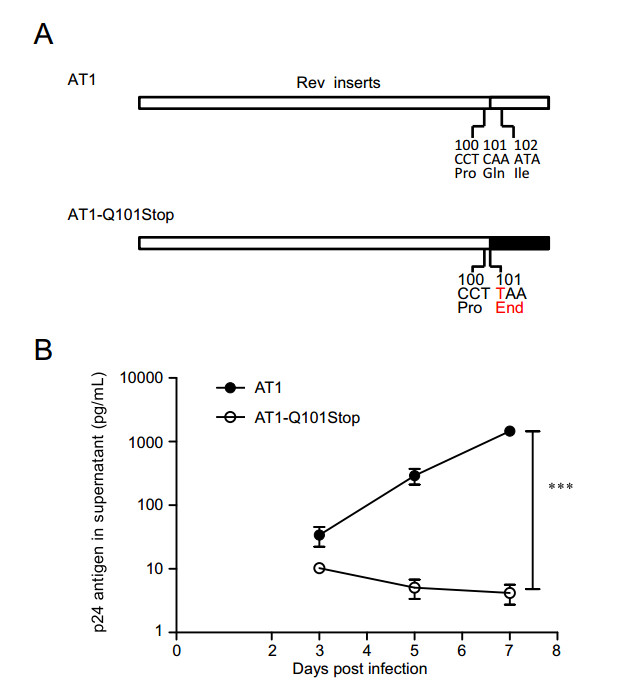
Figure 5. Replication capacities of clade B wild type and Rev Q101stop mutated virus in PM1 cells. A Diagrams of the mutation in clade B Rev. Red character suggests the mutated base or amino acid. B Replication capacities of virus mutants in PM1 cells. PM1 (7 × 105) cells were infected with 10 ng p24 antigen clade B wild type (AT1) or mutated virus (AT1-Q101Stop) virus in 1 mL total volume, then cells were washed twice by 10 mL PBS and seeded in 2 mL fresh medium. Supernatant (0.5 mL) was substituted by fresh medium at days 3, 5 and 7 post infection for p24 antigen quantification. Data are representative of at least three independent experiments. Statistical difference was presented as ***P < 0.001.
In order to measure the fitness change caused by the premature stop codon, growth competition between wild-type (AT2) and mutant viruses (AT1-Q101Stop) was performed in PM1 cells. PM1 cells were infected with these two viruses using the same p24 antigen amount, cells (5 × 105) were collected at days 3, 4, 5 and 6 post infection for flow cytometry analysis (Fig. 6A). PM1 cells coinfected with AT1 and AT2 were also collected as control for flow cytometry at days 3, 4, 5 and 6 after infection (Fig. 6B). Mutated virus AT1-Q101stop showed a very weak replication capacity compared with the wild-type virus at days 4, 5 and 6 (Fig. 6C, 6D). Compared with wild-type virus AT1, relative fitness of virus mutant (AT1- Q101Stop) was reduced by over 70% (Fig. 6E).
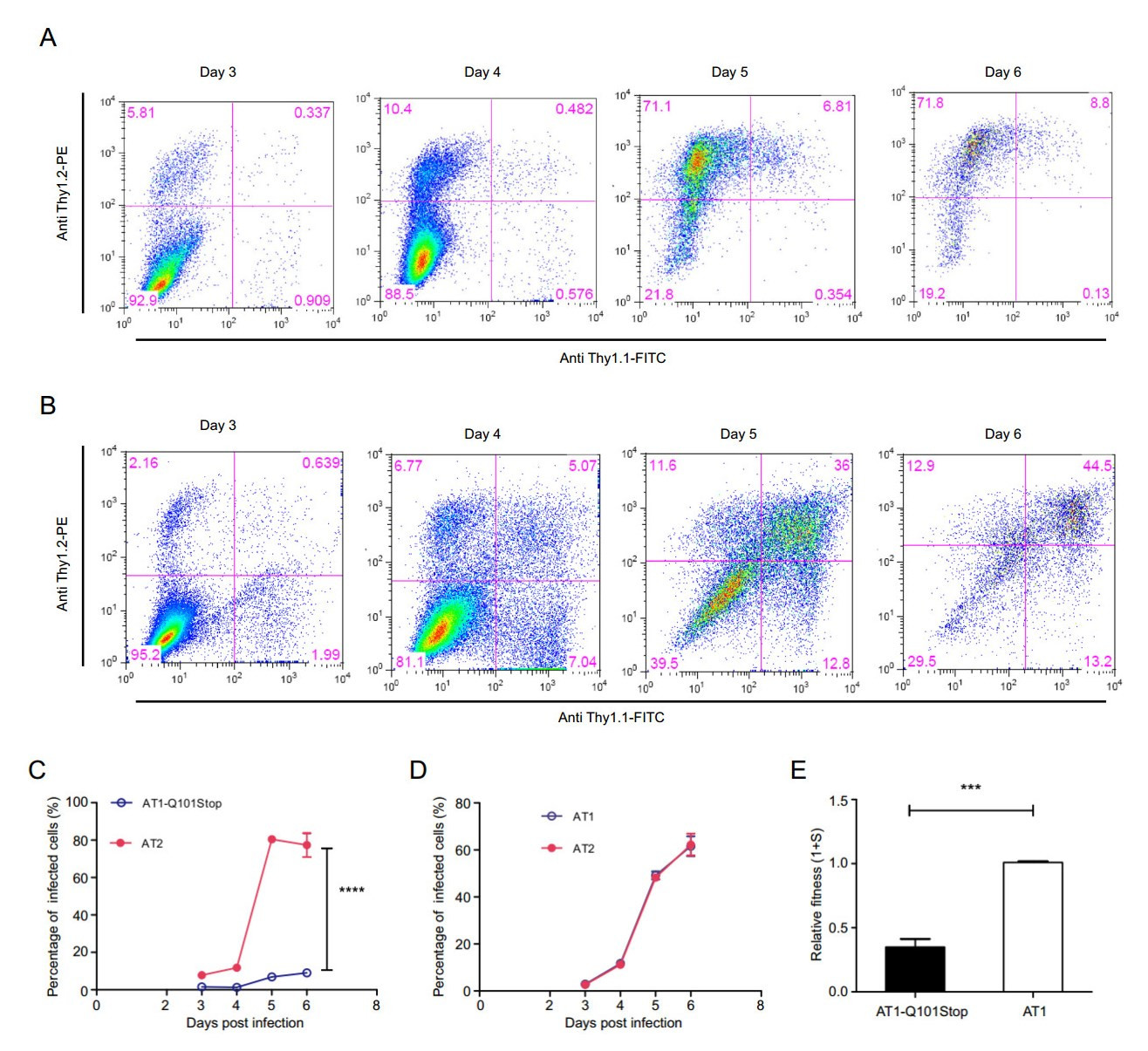
Figure 6. Growth competition assays of mutant virus versus wild-type virus of clade B. AT1-based mutant (300 ng p24) and AT2 (300 ng p24) were co-infected in PM1 cells (7 × 106). Cells (5 × 105) were harvested on days 3, 4, 5, and 6 post-infection, and stained by antiThy1.1 antibody, anti-Thy1.2 antibody respectively for flow cytometry analysis. The percentages of cells infected with virus mutant, wild-type virus and both viruses are labeled in the lower right, upper left and upper right quadrants, separately. AT1 (300 ng p24) and AT2 (300 ng p24) co-infected PM1 cells were collected as control. A Density plots of PM1 cells co-infected by AT1-Q101Stop and AT2 at days 3, 4, 5, and 6 post-infection. B Density plots of PM1 cells coinfected by AT1 and AT2 at days 3, 4, 5, and 6 post-infection. C AT1- Q101Stop versus AT2. D AT1 versus AT2. E Relative fitness of clade B virus mutants. Relative fitness of virus was calculated using the website tool (http://bis.urmc.rochester.edu/TFitness/FitnessTwo.aspx). Data are representative of at least three independent experiments. Statistical difference was presented as ***P < 0.001, ****P < 0.0001.
As Rev overlaps the transfer membrane (TM) of gp41, the introduced mutations may change the amino-acid of TM. For clade B or clade BC, the introduced mutations do not change the 282th or 289th AA of TM (Supplementary Figure S1), thus fitness reduction of virus mutants only can be attributed to the Rev mutations.
Clade BC/C Isolates have Higher Frequencies of Premature Stop Codon at Residue 101 Within Rev
Premature Stop Codon in CRF07_BC Rev Did Not Alter Rev Expression Level and Rev-RRE Activity
Substitution of Premature Stop Codon by Q in CRF07_BC Rev Did Not Alter Viral Replication Capacity
Mutation of Q101 to Stop Codon within Clade B Rev Impaired Rev Expression Level and Rev-RRE Activity
Mutation of Q101 to Stop Codon within Clade B Rev Weakened Viral Fitness
-
In this study, the frequency of premature stop codon within Rev of different HIV-1 clades was evaluated. It was found that this stop codon was present at a higher frequency (over 95%) in clade BC/C isolates. The premature stop codon at residue 101 within Rev leading to the truncation of 16 AAs, had been found to have no effect on replication of clade BC virus, yet efficiently reduced fitness of clade B virus. The fitness change is associated with the Rev-RRE activity.
Also, we found that premature stop codon was not only present in clade BC or C, but also in several circulating clades. To date, the disordered C-terminal portion of Rev including the NES that is required for recruitment of Crm1 and other host proteins that facilitate nuclear export of the Rev-RRE complex, was still unresolved (Rausch and Le Grice 2015). Whether truncated Rev affects its expression level and function in the different clades remains unclear. Previous research reported that C-terminal deletion (QILVESPTILE, residue 101-111) could decrease the phosphorylation level of Rev by 80%–95% (Malim et al. 1989). Recently, Dimattia et al. showed that truncation prevents filament formation, suggesting that the C-terminus affects the association process of Rev–Rev (DiMattia et al. 2016). Toth-Petroczy et al. predicted that the C-terminus folds back and interacts with residues in the NES (Toth-Petroczy et al. 2016). We confirmed that 16 AA truncation in C-terminal did impair Rev expression level and Rev-RRE activity in clade B, but had no effect on them in clade BC. A deletion of 7 AAs (QSQGVET) in clade B Rev may be associated with this deficiency (Fig. 1A), and Rev structure function by this deletion should be explored in the following research.
Rev-RRE activity is a key parameter by which the replication capacity of virus is measured. During this research, we found that substitution of Q by stop codon at residue 101 in clade B Rev decreased Rev expression and Rev-RRE activity and in consequence impaired viral replication. With attention to detail, viral fitness of clade B was reduced by over 70% by premature stop codon in comparison with wild-type virus. Similar to our results, Fernandes et al. employed deep mutational scanning of Rev and thereby demonstrated that the stop codon at residue 86 or beyond produces fit viruses using clade B NL4-3, in spite of that the fitness of the stop codon allele varies (Fernandes et al. 2016). On the other hand, it is found that premature stop codon within CRF07_BC Rev did not change Rev expression, Rev-RRE activity and replication capacity. However, whether Rev truncation in the other HIV-1 clades influences their replication capacities still remains unknown. Therefore, further research should be refined and expanded to reveal the corresponding mechanism with a deep understanding.
In conclusion, we for the first time disclosed that C-terminal truncation within clade BC Rev did not alter viral replication capacity, yet that truncation within clade B Rev impaired viral replication fitness. Our findings explicitly explain the reason why that the premature stop codon prevails in clade BC/C isolates but is rarely seen in clade B.
-
We appreciate the generous gifts of AT1 and AT2 plasmids from Dr. Carrie Dykes (The University of Rochester School of Medicine and Dentistry), as well as BH10-Rev expression plasmid and pCMV-Gag-Pol-RRE expression plasmid of clade B from Dr. Min Wei (Nankai University). Clade BC Gag-Pol-RRE expression vector was received from Dr. Jingwan Han (Beijing Institute of Microbiology and Epidemiology). CMV-EGFP plasmid was gifted from Dr. Lei Yu (Shenzhen Second People's Hospital). We would like to thank Hong Peng (The Division of Research of Virology and Immunology, National Center for AIDS/STD Control and Prevention) for the technical supports for this work. This study was partially funded by the National Natural Science Foundation of China (Grants 81872680 and 31600734), the Yong Scientific Research Foundation of NCAIDS/STD (Grant 2018AFQN002) and the SKID outstanding youth grant (2019SKLID402). Funding has no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
-
ZW and YS conceived and designed the experiments. ZW and XJ performed the experiments. ZW, XJ and KH analyzed the data. DL, YH and YS contributed reagents/materials/analysis tools. ZW, KH and LM wrote the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
Informed consent have been obtained from all participants and the studies have been approved by the Institutional Review Board of the National Center for AIDS/STD control and Prevention, China CDC.

 DownLoad:
DownLoad: