














HTML
-
In recent decades, the prevention and treatment of bacterial diseases have relied heavily on antibiotics. However, the negative effects of antibiotics are gradually emerging. For example, the emergence of a large number of drug-resistant bacteria has made the diseases of aquaculture animal more difficult to be controlled. There is an urgent need to find alternatives to antibiotics (Defoirdt et al. 2011). The viruses that infect bacteria, i.e., phages, are the most abundant microorganisms in marine environments (Suttle 2005; Liang et al. 2019) and one of the most important factors leading to bacterial death (Wang et al. 2019). In recent years, the use of phage to remove pathogenic bacteria in aquaculture has been proven to be a very promising disease prevention strategy (Hoshiba et al. 2010; Jun et al. 2014; Nishikawa et al. 2008).
Vibrios are ubiquitous in the marine and estuarine environments, accounting for more than 10% of the total aquatic culturable bacteria (Lin et al. 2018; Li et al. 2019; Yang et al. 2019). Many members of vibrios are pathogenic bacteria that can infect aquatic animals and induce foodborne diseases (Drake et al. 2007). Among them, Vibrio campbellii is an opportunistic pathogen widely distributed in aquaculture seawater. It can cause serious diseases of prawns, finfish, and mollusks, such as bacterial septicemia, luminescent vibriosis or red-leg disease, etc. (Defoirdt et al. 2007) and lead to huge losses in marine aquaculture (Halet et al. 2007).
To our knowledge, only seven lytic phages infecting V. campbellii have been isolated so far, but there is no comprehensive identification in both physiological and genetic levels. Among them, the genomes of five lytic phages OPB45, OPB48, OPB54, OPB58 and OPB66 isolated from cockle samples are not sequenced (Sangseedum et al. 2017), and the physiological characteristics of the other two phages VHS1 and SIO-2 are unknown (Baudoux et al. 2012; Khemayan et al. 2012). In addition, two temperate phages that can infect V. campbellii have been isolated, but they are not suitable for the treatment of pathogens in aquaculture (Lorenz et al. 2016).
Recently, a pathogenic bacterium V. campbellii 18 was isolated from diseased Kuruma prawn. Meanwhile, a lytic phage, which is capable of infecting V. campbellii 18, was also obtained. In order to evaluate the application potential of this phage in the treatment of V. campbellii infection in aquaculture, its physiological and genomic characteristics, potential metabolic gene functions and biological safety were analyzed comprehensively. Our study provides new insights into the species diversity of Vibrio phages and their application potential in treating aquaculture pathogens.
-
The V. campbellii 18 strain was kindly provided by Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, which is a pathogen isolated from diseased Kuruma prawn. It was cultured from a glycerol stock in RO medium (yeast extract 1 g, tryptone 1 g, sodium acetate 1 g, artificial seawater 1 L, pH 7.8–8.0) at 28 ℃ with shaking at 200 rpm for 12 h. The phage named vB_VcaS_HC was isolated using standard virus enrichment and double-layer agar method (Zhang and Jiao 2009). In detail, the samples for phage isolation were collected from the surface coastal seawater in Qingdao (36°17'42.56" N, 120°39'11.19" E) and filtered through 0.22 μm sterile microfilter. Then 10 mL filtrate was added to 50 mL V. campbellii 18 culture and incubated at 28 ℃ for 7 days in a shaker. Samples were taken from the mixed culture at an interval of 24 h. Each sample was filtered with 0.22 μm sterile microfilter and then mixed with V. campbellii 18 using the double-layer agar method. After overnight incubation at 28 ℃, single plaque was picked out and purified at least five times by the double-layer agar method.
-
Phage particles were prepared according to the conventional negative staining method (Liang et al. 2016). Briefly, phage stock suspension was placed on copper grids for about 30 min and then stained with 2% uranyl acetate for 3 min. The negatively stained phage was examined by H-7650 transmission electron microscope at an accelerating voltage of 80 kV.
-
To investigate the presence of lipid in vB_VcaS_HC, the phages were incubated with 0%, 2%, or 20% (v/v) chloroform with vigorously shaking for 1 min and then held at room temperature for 30 min. After centrifugation at 6000 × g for 5 min, the phages remaining in the supernatant were dropped onto V. campbellii 18 plate. Whether the phage capsid contains lipid was judged according to the emergence of plaques (Zhang and Jiao 2009).
-
For the one-step growth curve, the previously described method was used with minor modifications (Yang et al. 2017). Briefly, V. campbellii 18 was infected with the phage at a multiplicity of infection (MOI) of 0.01 and then incubated at 28 ℃ for 20 min. The unabsorbed phage particles were removed by centrifugation at 6000 × g for 10 min and the pellets were resuspended in 1 mL of RO medium. This process was repeated twice. Then 50 μL of the resuspended culture were transferred into 50 mL of RO medium and incubated at 28 ℃ with continuous shaking. Samples were collected at 15 min intervals (up to 5.5 h), then immediately diluted, and plated for phage titration with the double-agar plaque assay. Three replicates of the experiments were carried out in this study. The burst size was calculated as the ratio of the final titer of liberated phages to the initial count of infected bacterial cells.
-
In vitro lysis assays were performed as described previously with some modification (Yuan et al. 2019). In brief, phage suspensions were added to the cultures of V. campbellii 18 (OD600 = 0.3–0.4) at an MOI of 0.01. Then samples were taken from the mixture every 15 min for 5.5 h and serially diluted (tenfold each) with cold RO medium. The number of bacteria in each sample was quantified with the agar plate method. All assays were repeated in triplicates. The bacterial mortality caused by phage infection was calculated by comparing the number of bacteria in the phage infection group and the control group without the addition of phages.
-
Spot test (Zhang and Jiao 2009) was carried out on 14 Vibrio sp. strains listed in Table 1 to determine the host range of vB_VcaS_HC. Briefly, 1 mL of bacteria in the log phase and 4 mL of melted top agar (RO medium with 0.5% agar) were mixed and poured onto the bottom agar plates (RO medium with 1.5% agar). After a 10 min solidification at room temperature of 20–25 ℃, 20 μL of phage suspension (108 PFU/mL) were spotted onto the top agar. The plates were then incubated at 28 ℃ and examined for plaque formation daily for a week.
Strain Most closely related strain Origin Susceptibility VIB283 Vibrio alginolyticus Donated# — LMG 19158(T) Vibrio scophthalmi Collection — NBRC 104587(T) Vibrio azureus Collection — NBRC15630(T) Vibrio alginolyticus Collection — CAIM 1831(T) Vibrio alfacsensis Collection — CAIM 25(T) Vibrio gigantis Collection — E20121(T) Vibrio zhuhaiensis Collection — Vb 11.11(T) Vibrio atlanticus Collection — E20121(T) Vibrio zhuhaiensis Collection — R-3712(T) Vibrio chagasii Collection — 18 Vibrio campbellii Donated* + 20160303005-1 Vibrio parahemolyticus Donated* — VIB 72 Vibrio anguillarum Donated# — VIB 645 Vibrio harveyi Donated# — +: infected; -: uninfected.
*Donated by Professor Jie Huang, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences.
#Donated by Professor Xiaohua Zhang, Ocean University of China.Table 1. Host range of vB_VcaS_HC (+: infected; -:uninfected).
-
To test whether the phage replication is independent of the host RNA polymerase (RNAP), different concentrations of rifampicin were added to 5 mL of freshly grown host culture to inactivate host RNA polymerase (Takeru et al. 2017). Twenty min later, the host was infected with the phage at an MOI of 1.0 and incubated at 28 ℃ for 20 h. After centrifugation at 6000 × g for 5 min, the supernatant was collected for plaque assay by the double-layer agar method. Whether the phage can replicate independently can be inferred from the number of plaques on the plate.
-
The phage DNA was extracted according to the method described previously (Zhang and Jiao 2009). Briefly, the CsCl-purified phages were treated with DNase I (2 ng/L) and RNase A (2 ng/L) at room temperature for 2 h to reduce the host contamination. Then proteinase K (50 μg/mL), EDTA (20 mmol/L), and SDS (0.5% w/v) were added and proteins were digested for 3 h at 55 ℃. DNA was extracted by phenol: chloroform: isoamyl alcohol solution (25: 24: 1, v/v) and chloroform: isoamyl alcohol solution (24: 1, v/v). After being precipitated with 50% isopropanol (v/v) and centrifugation (12, 000 × g, 15 min, 4 ℃), the DNA pellet was air-dried and further resuspended in double distilled water. The genomic DNA was sequenced using the Illumina Hiseq × Ten (2 × 150 bp) by the Allwegene Company (Beijing, China). The assembly of quality-filtered reads was performed using SPAdes V.3.12.0 and the ORFs were predicted using the GeneMarks online server and ORF Finder (Ling et al. 2018). Translated ORFs were annotated by the BLASTP algorithm against the non-redundant (nr) protein database of the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/) with E-value < 0.001. tRNAs were detected with tRNAscan-SE (Liang et al. 2016). The putative promoter sequences were predicted using the promoters online analysis tool Softberry (Carlota et al. 2016) while the phage termini and DNA-packing strategy were identified using PhageTerm (Garneau et al. 2017). The prediction of bacterial virulence genes, antibiotic resistance genes were performed with the VirulenceFinder (Joensen et al. 2014) and ResFinder server (Ea et al. 2012), respectively. The complete genome sequence of the phage vB_VcaS_HC was deposited at GenBank under accession number MK559459.1.
-
To test whether the dsDNA genome of vB_VcaS_HC is circular, a PCR reaction was conducted using the designed primer hc-F (5'-CCTACAAGGTAGTCAAGATA-3') and hc-R (5'-GTGCGAACTACAGCTAGAC-3') extending outward from the two termini of the "linear" phage genome respectively. Thirty microliters of the PCR reaction mix contained 2 × Taq Master Mix (Vazyme, China), 0.1 μmol/L of the forward and reverse primer each, and 50 ng of phage genomic DNA. Cycling conditions were 95 ℃ for 3 min, followed by 30 cycles at 95 ℃ for 30 s, 47 ℃ for 30 s and 72 ℃ for 2 min, with a final elongation step at 72 ℃ for 10 min. If the phage genome is circular, the PCR product should be 1, 235 bp in length and has a sequence consistent with the phage genome.
-
For lysogeny test, the previously described method was used with minor modifications (Khemayan et al. 2006). Briefly, the overnight culture of the host V. campbellii 18 (100 μL) was spread on RO plates. Then 10 μL of phage suspension (108 PFU/mL) was dropped onto the agar. After 12 h of incubation, big plaques appeared on the plate, meanwhile, several bacteria colonies grew inside the plaques. Then these bacterial colonies were picked out and purified by streak plate method. The susceptibility of these bacteria to phage infection was determined by the doublelayer agar method. We speculate that some of the bacteria that can resist phage infection may be lysogenized by phage vB_VcaS_HC. To test this, we extracted the genomes of all the phage-resistant bacteria and detected whether they contained fragments of phage genome by PCR analysis using a pair of specific primers designed for the phage capsid protein gene (ORF 44). The primers are 044F (5'-GTGGGTTTAGTGTGATGCGA-3') and 044R (5'-CTAGTGTTGTGATCGATACC-3'). The PCR conditions are as follows, thirty microliters of the PCR reaction mix contained 1 μL Phanta Max Super-Fidelity DNA Polymerase (Vazyme, China), 2 × Phanta Max Buffer, 0.1 μmol/L of each forward and reverse primer, and 0.2 μL of template. Cycling conditions were 95 ℃ preheating for 3 min, followed by 30 cycles of denaturation at 95 ℃ for 30 s, annealing at 48 ℃ for 30 s and extension at 72 ℃ for 1 min, with a final elongation step at 72 ℃ for 10 min. Meanwhile, the phage genomic DNA was used as template of the positive control and the genomic DNA extracted from the wild type V. campbellii 18 were used as template of the negative control. The PCR amplification products were detected by agarose gel electrophoresis and analyzed by DNA sequencing.
-
The most similar phages to the phage vB_VcaS_HC were identified using BLASTn (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Their sequences were downloaded from GenBank and used for further comparative analysis. Phage genome visualization and the alignment of different phage genomes were performed using CGView (Grant and Stothard 2008). CoreGene3.5 was used for pairwise comparisons with the default "75" score stringency (Turner et al. 2013). The amino acid sequences of DNA terminase and RecA of vB_VcaS_HC were used to construct phylogenetic trees using the neighbor-joining method in MEGA 7.0 and ClustalW. Meanwhile, the phage genome was uploaded to the ViPTree (https://www.genome.jp/viptree) to generate a proteomic tree.
Phage Isolation
Observation of Phage Morphology by Transmission Electron Microscope
Detection of Phage Lipids
One-Step Growth Curve
Bactericidal Activity
Host Range Analysis
Analysis of Phage Replication
Phage DNA Isolation, Genome Sequencing and Analysis
Determination of Genome Type (Linear or Circular)
Lysogeny Test
Comparative Genomics and Phylogenetic Analysis
-
TEM examination revealed that the phage vB_VcaS_HC had an isometric head with 81 ± 2 nm in diameter and a non-contractile tail with 206 ± 6 nm in length, morphologically belonging to the Siphoviridae family (Fig. 1A). The measured phage tail length is almost the same as the predicted length (207 nm) according to the length of tape measure protein (ORF37) in the phage genome (Katsura 1987). When treated with chloroform, the infectious activity of the phage remained well, indicating it is a lipidfree phage in the capsid (Fig. 1B).
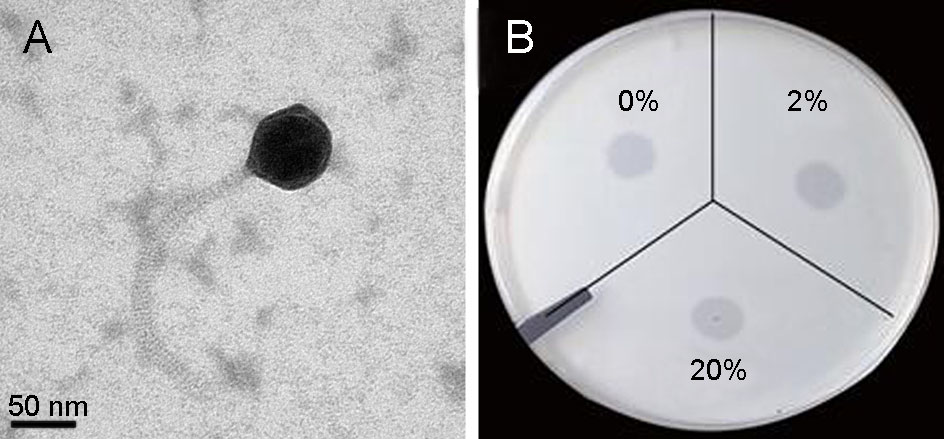
Figure 1. A Transmission Electron micrograph of phage vB_VcaS_HC. Scale bar represents 50 nm. B Chloroform sensitivity of vB_VcaS_HC infecting Vibrio campbellii 18.
-
One-step growth experiment revealed that the phage vB_VcaS_HC had a latent period of 1.5 h and a burst size of 172 PFU/cell (Fig. 2A). The phage infection caused a sharp decrease in the number of host V. campbellii within 3 h (Fig. 2B). In 5.5 h, the host mortality reached up to 96%. Among the 14 Vibrio strains tested, none except V. campbellii 18 was susceptible to phage infection (Table 1). To test whether vB_VcaS_HC replication was independent of the host, vB_VcaS_HC was treated with rifampicin. The results showed that the phage infection was completely abolished when the host RNAP was inhibited by rifampicin (Supplementary Table S1), suggesting that the transcription of phage vB_VcaS_HC must depend on RNAP encoded by host (Takeru et al. 2017).
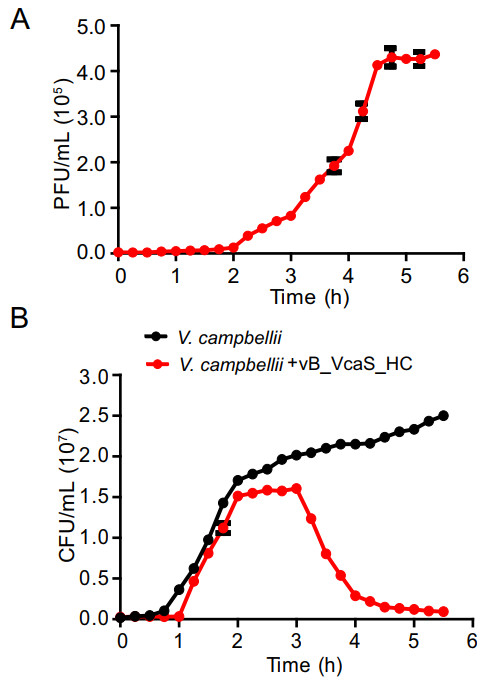
Figure 2. A One-step growth curve of phage vB_VcaS_HC. B The growth curves of the phage infected and uninfected V. campbellii 18. Values represent the mean ± SD (n = 3).
-
The phage vB_VcaS_HC has a circular dsDNA genome with the length of 81, 566 bp. The G + C content is 47.6%. The phage genome contains 194 promoters. No tRNA sequence was found in the phage genome, which indicates that the phage transcription is completely reliant on the host tRNA and its machinery (Ceyssens et al. 2006). The termini analysis revealed that the phage vB_VcaS_HC may display a headful (pac) packaging mechanism and has redundant ends to circularize the phage genome through recombination (Garneau et al. 2017).
The genome of this phage contains 121 ORFs, all of which are transcribed in the same direction. Of the 121 ORFs, only 35 ORFs are assigned putative functions based on their amino acid sequence homology with known proteins. About 71% of the ORFs, i.e., 86 ORFs, are annotated as proteins with unknown functions, 8 ORFs of which have no homology with any protein in NCBI database. All the functionally-annotated ORFs in the genome were listed in Supplementary Table S2.
In the phage genome, no bacterial virulence or drugresistance gene was detected. Fourteen ORFs encode proteins related to structural formation, such as coil-containing protein, neck protein, major capsid protein, major tail protein, tail length tape measure protein, head completion adaptor and portal protein, etc. The portal protein encoded by ORF118 is a molecular motor participating in DNA packing (Moore and Prevelige 2002). In addition, 10 ORFs encode proteins related to DNA replication, including terminase large subunit, ribonuclease, DNA methyltransferase, DNA helicase, DNA polymerase I, DNA-binding domain protein, etc. These proteins can be divided into the early DNA replication proteins, DNA replication and repair proteins, and DNA packaging related proteins (Marina et al. 2017). The early DNA replication proteins mainly refer to DNA helicase encoded by ORF20 and ORF24, whose function is to unwind double-stranded DNA (Wu 2012). The DNA replication and repair proteins include the DNA polymerases encoded by ORF113 and ORF119, whose function is to elongate the primer during genome replication. The DNA packaging related proteins mainly refer to the terminase enzymes encoded by ORF8, which is responsible for slicing the dsDNA into the final genomesized sequence and then helps package the entire genome into a pre-assembled empty capsid (Hamdi et al. 2017).
Moreover, 10 ORFs were annotated as phage auxiliary metabolic genes encoding rubredoxin-type fold protein, PPDK and transporter, etc. Interestingly, although this phage is characterized as a lytic phage, whereas a lysogeny-related gene, i.e., ORF17 encoding recombinase (RecA), is present in the phage genome, but in fact our study did not observe the occurrence of lysogeny.
BLASTn analysis using the complete genome sequence revealed the highest sequence similarity of the phage vB_VcaS_HC to the unclassified Vibrio phage 1 (JF713456.1, 81.82% sequence identity), followed by the unclassified Vibrio phage Ares1 (MG720309.1, 80.86% sequence identity), Vibrio phage vB_VhaS-a (KX198615.1, 69.66% sequence identity) and Vibrio phage SIO-2 (HQ316604.1, 66.58% sequence identity). Core genes are common genes between sets of genomes and have been used to get a better understanding of viral evolutionary complexity (Yutin and Koonin 2012). We used coregene3.5 for pairwise aligning of vB_VcaS_HC to phage genomes of VHS1 and SIO-2. As can be seen in Table 2 and Fig. 3, vB_VcaS_HC shares a high gene correlation with those two phages.
Phage VB_VcaS_HC VHS1 SIO-2 Genome length (bp) 81566 81509 80598 GC content (%) 47.6 46.87 45 t RNA No No No ORF 121 125 116 Circular or linear Circular Circular Circular Classification Siphovirus Siphovirus Siphovirus Origin China Thailand Northern coastal Pacific Ocean CoreGenes3.5 Correlation — 88% 68.7% Table 2. Basic genome characteristics of the phage vB_VcaS_HC, VHS1 and SIO-2.
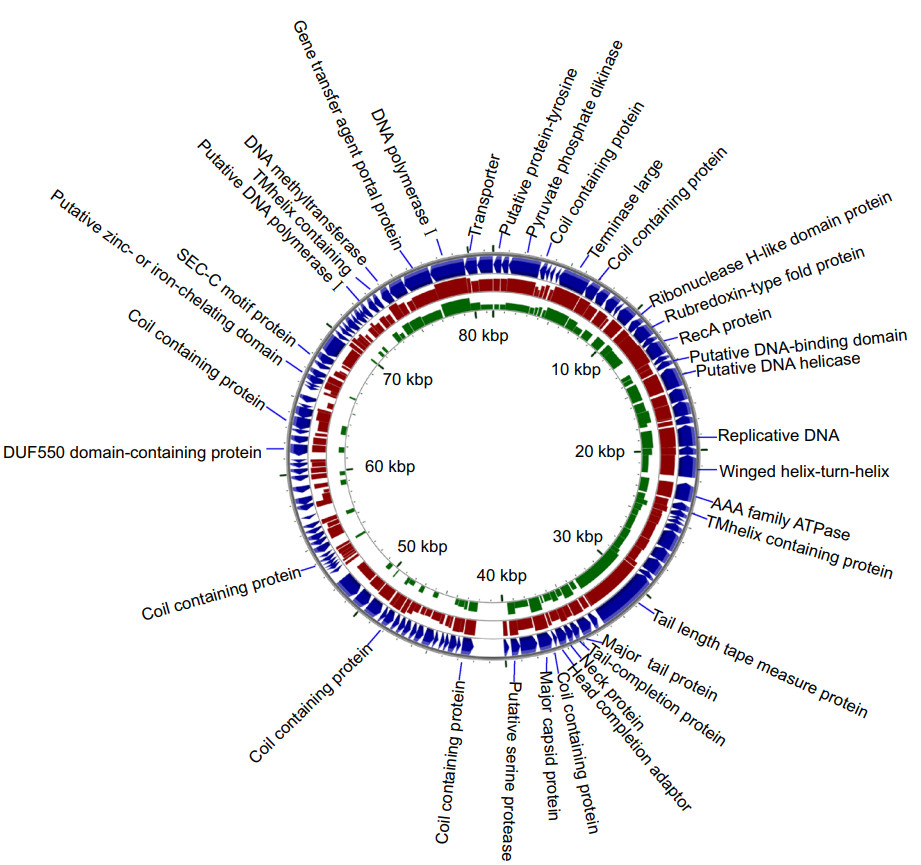
Figure 3. Alignment of the phage genome of vB_VcaS_HC (the blue outermost ring; Accession No. MK559459.1) against that of Vibrio phage 1 (red ring; Accession No. JF713456.1) and Vibrio phage SIO-2 (green ring; Accession No. HQ316604.1) using blastx (minimum E-value: 0.00001). The height of each arc in the BLAST results rings is proportional to the percent identity of the hit.
To further classify the phage vB_VcaS_HC, phylogenetic trees based on phage proteome, DNA terminase and RecA amino acid sequence were established respectively (Fig. 4). The phylogenetic position of phage vB_VcaS_HC shows that it has the highest homology with the phage VHS1, which was unclassified by ICTV (International Committee on Taxonomy of Viruses) yet. Moreover, phage vB_VcaS_HC has a close evolutionary relationship with the Stenotrophomonas phage vB_SmaS_DLP_5 (Fig. 4), which is affiliated to Delepquintavirus, Siphoviridae, Caudovirales (Peters and Dennis 2018).
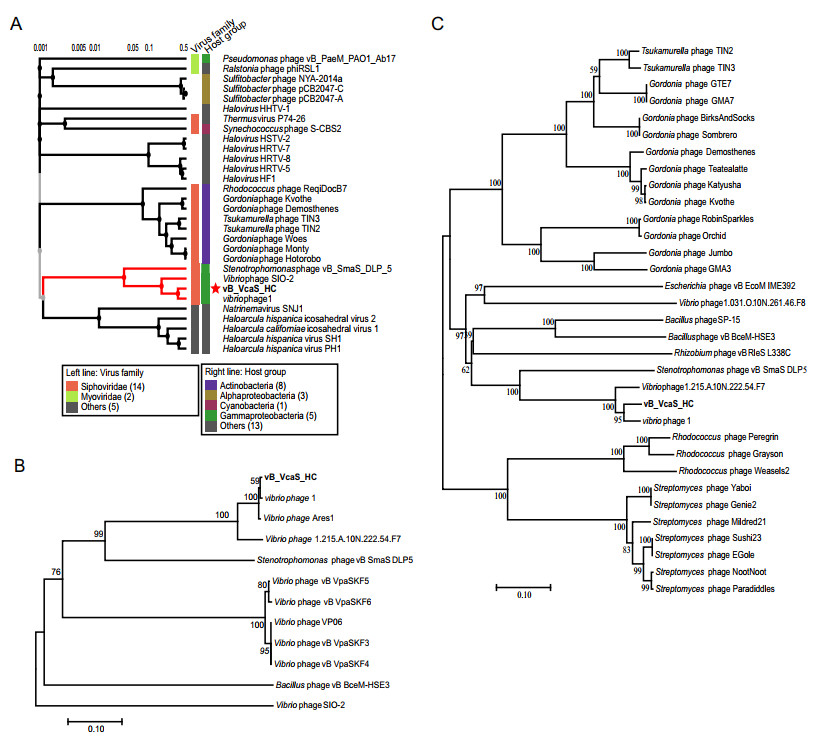
Figure 4. Phylogenetic relationships between vB_VcaS_HC and other phages. A The viral proteomic tree including vB_VcaS_HC and other closest relative phages constructed using the ViPTree. B, C Neighborjoining phylogenetic trees of the RecA protein and DNA terminase, respectively. All reference sequences were collected from the NCBI database. The trees were constructed based on the MUSCLE alignment of MEGA 7.0. The bootstrap values were based on 10, 000 replicates.
Morphological and Structural Characteristics of the Phage vB_VcaS_HC
Phage Infection Characteristics
Phage Genome Analysis and Phylogenetic Position of the Phage vB_VcaS_HC
-
This study reported a lytic vibriophage vB_VcaS_HC infecting the prawn pathogen V. campbellii. It can cause up to 96% mortality of the pathogen host in 5.5 h, showing a good application potential in the treatment of pathogen V. campbellii 18. In additioon, its host range is very narrow, therefore when it is used in the treatment of pathogens, it might not infect other potentially harmless bacteria.
Whole genome analysis reveals that the phage vB_VcaS_HC contains a circular dsDNA genome. Seventy-one percent of the genes in the genome are unknown in function. In addition to the ORFs encoding structure and DNA replication related proteins in the phage genome, there are ten auxiliary metabolic genes encoding rubredoxin-type fold protein, PPDK and transporter, etc., which may be involved in host metabolism during infection (Ceyssens et al. 2006). For example, rubredoxin-type fold protein (ORF15) is the simplest type of iron-sulfur protein ubiquitous in life systems. It can act as a small electron transfer protein participating in many biocatalytic reactions to generate energy or act as an oxygen oxidoreductase to protect bacterial cells by preventing the formation of reactive oxygen species (ROS) under microaerophilic conditions (Wildschut et al. 2006). The PPDK exists in bacteria, protists and plants, but has never been found in phages. It catalyzes the reversible interconversion of phosphoenolpyruvate and pyruvate, which plays an important role in the Embden-Meyerhof-Parnas (EMP) pathway, the main pathway of glucose degradation and energy generation. In archaea, the PPDK and related enzymes participate in the regulation of carbohydrate metabolism and allow its adaptation to different nutritional conditions (Tjaden et al. 2006). We propose that when the phage vB_VcaS_HC carrying PPDK gene infects the host cell, the expression of phage-derived PPDK may participate in the carbohydrate metabolism of the host to produce more ATP, thereby providing more energy for the assembly and propagation of phage progeny.
Interestingly, although the phage vB_VcaS_HC showed a strong lytic ability to kill the pathogen, we found a potential lysogeny-related gene, i.e. the recA gene (ORF17), in the phage genome. For bacteriophage γ, recA gene is expressed conditionally by its host Escherichia coli K12 in response to DNA damage, its expression product then triggers a cascade of gene activation, leading to the excision of prophage genome from the host genome. The phage genome then replicates in the host cell, resulting in cell lysis. In that way, RecA participate in the transition of phage from lysogeny to lysis (Morelli et al. 2009). In the genome of another Vibrio phage VHS1, the recA gene has also been observed, and the phage VHS1 was verified that could switch to lysogenic status from lytic status (Khemayan et al. 2006). Here, we analyzed the amino acid sequence similarity of the three RecA proteins from the phage vB_VcaS_HC, E. coli K12 and phage VHS1 and found that the RecA proteins of vB_VcaS_HC and VHS1 have a very high homology (100% query cover, 96.93% sequence identity) (Supplementary Figure S1), suggesting that the phage vB_VcaS_HC is likely to be able to transform into a lysogenic state like the phage VHS1 (Khemayan et al. 2006). However, both the two RecA proteins of the phage vB_VcaS_HC and VHS1 have a much lower similarity with that of the host of bacteriophage λ (vB_VcaS_HC vs E. coli K12: 62% query cover, 34.65% sequence identity; VHS1 vs E. coli K12: 85% query cover, 31.45% sequence identity). The lysogeny refers to the state of a host bacterium that has incorporated a phage into its own genome (Zhang et al. 2011). The lysogenized bacteria will not be infected by the same type of phages again. Usually when a phage infects a bacterium, it can either destroy the bacterium or be incorporated in the bacterial genome in a lysogenic state (Huang et al. 2010; Zhang et al. 2009).
To verify whether the phage vB_VcaS_HC can really turn into lysogenic state during infection, we picked out all the bacterial colonies that grew inside the dot plaques (Supplementary Figure S2) formed by phage infection on the plate covered with host bacteria and found that these bacteria were resistant to phage infection. Then through PCR assay, we confirmed that the genomes of these phageresistant bacteria do not contain the phage marker gene (i.e., the major capsid protein gene of phage vB_VcaS_HC). This precludes the possibility of superinfection immunity and prophage repression. Meanwhile, it indicates that at least under our experimental conditions, the phage vB_VcaS_HC did not become lysogenic. However, since the transition of phage between lytic status and lysogeny is usually determined by a cascade of genes activition (Morelli et al. 2009), besides recA in phage vB_VcaS_HC, although no other potential lysogenic genes (e.g., integrase, excisionase, transposase) were observed, we cannot exclude the possibility that one or some unknown genes may be involved in the switch between lytic status and lysogeny, as a large portion of the phage genes are of unknown function. Further study is still needed to reveal the actual function of RecA in the phage vB_VcaS_HC. Moreover, it is unclear whether the host genes related to phage infection cycle have been mutated, as bacteria usually gain resistance to phage through genetic mutation (Huang et al. 2010; Zhang et al. 2009).
Comparative genome analysis reveals that the phage vB_VcaS_HC is a novel species unknown before, as its highest genome similarity to the three closest virophages (i.e., Vibrio phage Ares1, vB_VhaS-a, and SIO-2) is only about 81%, much lower than the threshold (i.e., 95% genome sequence identity) for demarcation of new species (Evelien and Rodney 2017). Among these phages similar to vB_VcaS_HC in genome, two published phages Vibrio phage 1 (VHS1) and Vibrio phage SIO-2 (SIO-2) infecting V. campbellii were aligned using the CGView Server. VHS1 was isolated from a black tiger shrimp-rearing pond, which can infect V. campbellii 1114GL (Ke et al. 2017; Khemayan et al. 2012). SIO-2 was isolated from the surface water of the Northern coastal Pacific Ocean, which can infect V. campbellii ATCC 25920 and ATCC BAA-1116 (Baudoux et al. 2012). We found that our phage vB_VcaS_HC and the phage of VHS1 and SIO-2 are all siphovirus. Meanwhile, they all contain a dsDNA circular genome with similar length (81, 566, 81, 509, and 80, 598 bp, respectively) and oriented in the same direction. Comparative genomic analysis implied that these three phages have the same genome architecture and have high similarity in the sequence of their translation products (Fig. 3).
Moreover, the phylogenetic analysis based on phage proteome, DNA terminase and RecA amino acid sequences further confirms the phage vB_VcaS_HC as a novel species. Since the phage vB_VcaS_HC share 69.66% identity with the Stenotrophomonas phage vB_SmaS_DLP_5, which is affiliated to Delepquintavirus, Siphoviridae, Caudovirales (Peters and Dennis 2018), and 95% and 50% genomic similarity are usually regarded as the threshold to divide new species and genus, respectively (Evelien and Rodney 2017), hence vB_VcaS_HC is considered as a new species of Delepquintavirus.
Taken together, the vibriophage vB_VcaS_HC has a strong lytic ability against pathogenic bacteria and a high infection specificity. Although this phage contains a lysogeny-related gene, but actually lysogeny did not occur in our study. In addition, the phage genome does not contain any virulence or antimicrobial resistance genes. All these render this phage a promising candidate for the treatment of pathogen infections in aquaculture. Interestingly, the phage genome also contains several auxiliary metabolic genes encoding rubredoxin-type fold protein, PPDK and transporter, etc., among which the gene encoding pyruvate phosphate dikinase has never been found in other phages. At present, due to the lack of sufficient isolated strains, our knowledge of vibriophage is still very limited. Isolation and comprehensive identification of vibriophage is a very important work in the future. This is of great significance for us to develop phage cocktails that can replace antibiotics to effectively control aquaculture pathogens on the basis of fully understanding their diversity and different infection strategies.
-
This work was supported by the open task of Qingdao National Laboratory for Marine Science and Technology (QNLM2016ORP0311), the NSFC projects (41876174, U1906216), the Senior User Project of RV KEXUE (KEXUE2019GZ03) supported by Center for Ocean Mega-Science, Chinese Academy of Sciences, the DICP & QIBEBT (DICP & QIBEBT UN20 1803), the QIBEBT (QIBEBT ZZBS 201805), Dalian National Laboratory For Clean Energy (DNL), CAS, and Central Public-interest Scientific Institution Basal Research Fund, CAFS (No. 2017HY-ZD1002).
-
YZ designed the experiments and revised the manuscript; ZW, JZ, LW, GX and JH helped to analyze the data and revise the manuscript; CL performed the experiments, analyzed the data and wrote the manuscripts. YZ checked and finalized the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest to this work.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.

 DownLoad:
DownLoad: