














HTML
-
The continual spread of African swine fever (ASF) in European countries, African countries, Russia and further in China, has posed a severe threat to the global swine industry and food security (Dixon et al. 2019; Simulundu et al. 2017). In China, pork and pork products are important source of meat, providing people with the main source of protein and energy. However, ASF has been spreading rapidly in China ever since its emerging in 2018, severely destroying Chinese swine industry and food safety (Ge et al. 2018; Wang et al. 2018). As a double strand DNA virus belonging to family Asfarviridae, genus Asfivirus, ASFV is the causative agent of ASF, resulting in symptoms including high fever, hemorrhage and ataxia etc. (Karger et al. 2019; Mazur-Panasiuk et al. 2019; Galindo et al. 2017). In swine industry, inactivated vaccine has not been completely successful in protecting from ASFV infection (Teklue et al. 2019; Malogolovkin et al. 2019; Gaudreault et al. 2019). In addition, adenovirus-based vaccines or subunit vaccines play very little role in protection against the infection of virulent strains of ASFV (Sanchez et al. 2019). In this case, quarantine of ASF is particularly important, especially in ASF free regions and states.Therefore, it is of great urgent to develop a rapid and reliable method for quick detection of ASFV.
As a kind of well-developed technique, PCR is a recommended method for determination and confirmation of suspected cases in clinical diagnosis. PCR techniques are especially useful for the detection of the samples that are unsuitable for viral isolation due to putrefaction. A number of conventional PCR and qPCR assays have been established in previous studies of ASFV DNA detection (Aguero et al. 2003; Basto et al. 2006; Fernandez-Pinero et al. 2013; Wang A et al. 2019). The traditional PCR assays with high sensitivity and specificity have been wildly used in routine laboratory diagnosis. However, some drawbacks in sensitivity, specificity, simplicity and expenditure still limit the application of traditional PCR assays for further research and clinical purpose.
The CRISPR-lwCas13a is an ortholog of Cas13a from Leptotrichia wadei. It can be reprogrammed with CRISPR RNA (crRNA) to specifically target the examined RNA (Abudayyeh et al. 2016). Afterwards, the collateral-cleavage effect of CRISPR-lwCas13a is activated to degrade labeled RNA in vitro (Smargon et al. 2017; East-Seletsky et al. 2016). The RPA-CRISPR detection method is also called specific high-sensitivity enzymatic reporter unlocking (SHERLOCK). It is an in vitro nucleic acid detection platform with a single copy sensitivity based on RPA and CRISPR-mediated collateral cleavage of a reporter RNA (Gootenberg et al. 2017; Piepenburg et al. 2006). Since RNA reporter is the cleavage substrate of cas13a, the RPA amplicon need to be further transcribed into target RNA to activate the cleavage reaction. Notably, the copy number of crRNA target region will be amplified for the second time by DNA transcription after RPA amplification. Therefore, Cas13a is suitable for detecting samples with extremely low viral load in perspective of sensitivity.
In combination with heating unextracted diagnostic samples to obliterate nucleases (HUDSON) treatment, RPA-CRISPR can rapidly detect the viral DNA/RNA in body fluids obtained from infected animals without the time-consuming process of DNA extraction (Myhrvold et al. 2018). When it is used with a lateral flow strip, the application of RPA-CRISPR may be further expanded for field diagnosis (Myhrvold et al. 2018). Thus, RPACRISPR will greatly meet the requirements of food inspection and disease control in swine industry. In this study, an RPA-CRISPR-based highly sensitive diagnosis method was developed for rapid detection of ASFV for both laboratory and clinical purpose.
-
The CRISPR-lwCas13a prokaryotic expression vector pC013-Twinstrep-SUMO-huLwCas13a was obtained from Addgene (Addgene plasmid # 90, 097; http://n2t.net /addgene: 90, 097; RRID: Addgene_90097) (Gootenberg et al. 2017). A pMD-18 T-P72 plasmid was constructed by inserting complete coding sequence of ASFV P72 gene into a pMD-18 T vector. Both PCV-2 ZJ/C and PCV-2 SH are commercial vaccine strains of porcine circovirus type 2 (PCV-2) that are wildly used in China (Chen et al. 2018). Classical swine fever virus (CSFV)-C strain is an attenuated live vaccine strain originated from a virulent strain (CSFV-Shimen strain) after passages in rabbits (Luo et al. 2014). PRRSV-JAX1 and PRRSV-R98 are commercial vaccine strains of porcine reproductive and respiratory syndrome virus (PRRSV) used in China (Han et al. 2017). PRV Bartha-K61 is the first attenuated live vaccine strain of pseudorabies virus (PRV) (Freuling et al. 2017). PRV HB-98 is a double deletion modified live vaccine strain of PRV, which is wildly used in China. A recombinant of chimeric transmissible gastroenteritis virus (TGEV) and porcine epidemic diarrhea virus (PEDV) was also purchased as a commercial vaccine. Vaccines of PCV-2 ZJ/C, PRV Bartha-K61, PRRSV-R98, CSFV-C and TGEV + PEDV were purchased from Ringpu Biological Pharmaceutical Co., Ltd, Baoding, Hebei, China. Vaccine of PCV-2 SH was purchased from Jiangsu Nannong Hi-Tech Co., Ltd, Wuxi, Jiangsu, China. Vaccines of PRV HB-98 and PRRSV-JAX1 were purchased from Wuhan Keqian Biology Co., Ltd, Wuhan, Hubei, China. All of the DNA samples isolated from the ASFV positive and ASFV negative blood were obtained from Hubei Center for Animal Disease Control and Prevention, Wuhan, Hubei, China.
-
The crRNA template was an 84 nt single strand DNA (ssDNA) consisting of T7 recognizing sequence, repeat sequence and spacer sequence (Supplementary Table S1) (Shmakov et al. 2017). For preparation of crRNA, double strand DNA (dsDNA) was amplified by using the template of 84 nt ssDNA and its flanking primers (Supplementary Table S1). After DNA extraction with Gel Extraction Kit (Omega), dsDNA product of crRNA was used for RNA transcription with a HiScribeTM T7 High Yield RNA Synthesis Kit (New England BioLabs). The transcription product was treated with DNase I at 37 ℃ for 1 h to degrade the dsDNA template. The crRNA final product was extracted with TRIZol Reagent (Invitrogen) and was stored at - 80 ℃ for further use.
-
E. coli. BL21 (DE3) competent cells were transformed with the CRISPR-lwCas13a prokaryotic expression vector pC013-Twinstrep-SUMO-huLwCas13a and were cultured in a 4 L of LB broth (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl, Sigma) at 37 ℃ until an OD600 of 0.6. Subsequently, protein expression was induced by supplementation of 500 μmol/L IPTG (Invitrogen) at 18 ℃ for 16 h. After centrifugation, cells were collected and resuspended in lysis buffer (20 mmol/L Tris–HCl, 500 mmol/L NaCl, pH 8.0) containing 1× protease inhibitor (cOmpleteTM Protease Inhibitor Cocktail, Roche). Cell lysate was obtained by using a nano homogenize machine (ATS Engineering INC) under pressure of 600 bar and was centrifuged at 13, 000 ×g for 50 min. After it was filtered through a 0.22 lm filter (Millipore), the supernatant was purified by a HisTrap FF Column (GE Healthcare Life Sciences) in an NGC Quest 100 protein purification system (Bio-Rad). The eluted fragments were pooled and then digested by SUMO protease (Invitrogen) at 4 ℃ overnight. The digestion product was further purified by a gel filtration column (Superdex® 200, GE Healthcare Life Sciences) and the purified product was analyzed by SDSPAGE. The buffer containing corrected fragments was replaced with storage buffer (600 mmol/L NaCl, 50 mmol/L Tris–HCl, 5% glycerol, pH 7.5) and the corrected fragments were frozen at -80 ℃ for further use.
-
A pair of primers targeting P72 gene of ASFV genomic DNA were obtained by pre-screening according to instruction of RPA kit (TwistAmp® Liquid Basic, TwistDx). The RPA forward primer consisted of a T7 transcription region and target region (5'-TAATA CGACTCACTATAGGGACATTAAAAATGTGAACAA A-3'). The reverse primer merely consisted of a target region (5'-CTCTAAAGGTGTTTGGTTGTCCCAGTCAT AT-3'). In a 50 μL total reaction tube, regents consisted of 25 μL 2 × Reaction Buffer, 5 μL 10 × Basic E-mix, 2.5 μL 20 × Core Reaction Mix (TwistAmp® Liquid Basic, TwistDx), 3.6 μL dNTPs (25 mmol/L), 2.5 μL forward primer (100 μmol/L), 2.5 μL reverse primer (100 μmol/L), 0.5 μL RNase inhibitor (New England BioLabs), 1 μL MgCl2 (250 mmol/L), 2.5 μL NTP Buffer, 0.5 μL T7 Mix (New England Biolabs), 1 μL synthesized RNA reporter (10 μmol/L) (Sangon Biotech) (5'-FAMUUUUU-BHQ-3'), 81.3 ng CRISPR-lwCas13a protein, 131.5 ng crRNA, 2.5 μL MgOAc (280 mmol/L) and DNA template. After vortex and centrifugation, reaction tubes were incubated at 37 ℃ in a CFX96 Real-Time System (Bio-Rad) with FAM fluorescent signal detected every minute. The fluorescence intensities difference between samples and blank control in the final cycle were calculated and analyzed by GraphPad Prism 8.
-
Acrylamide denaturing electrophoresis was performed to analyze the RNA reporter cleavage. Briefly, 100 μmol synthesized RNA reporter (Sangon Biotech) (5'-FAMUUUUU) was mixed with 81.3 ng CRISPR-lwCas13a protein, 131.5 ng crRNA, 1 μL MgCl2 (250 mmol/L) and 10 ng ASFV target RNA in a 50 μL total reaction at 37 ℃ for 30 min. The target RNA was obtained by RNA transcription by using RPA amplicon as template. RNase I was mixed with RNA reporter as positive control. The 50 μL reaction mixtures lacking components crRNA, Cas13a or target RNA were set as negative controls. The samples mentioned above were analyzed by 40% Acrylamide denaturing electrophoresis. The fluorescent bands were visualized under a Gel Doc XR + Gel imaging system (Bio-rad).
-
In HUDSON treatment, Tris (2-carboxyethyl) phosphine hydrochloride (TCEP) (Sigma) at a final concentration of 100 mmol/L and EDTA (Sigma) at a final concentration of 1 mmol/L were added into a single sample solution containing ASFV viral DNA. Inactivation at 50 ℃ for 20 min was followed by lysis at 95 ℃ for 5 min on a thermocycler. Afterwards, sample solutions were directly added into RPA-CRISPR collateral reaction system for DNA detection.
-
Viral DNA and RNA were extracted from vaccines and ASFV positive specimens by using a TIANamp Virus DNA/RNA Kit (Tiangen Biotech). Viral RNA was reverse transcribed into cDNA with an FSQ 201 ReverTra Ace kit (TOYOBO). Forward primer (5'-CGCCGTTTACACGCT TGTAG-3'), reverse primer (5'-AACCAAGTTTCGGTA CGCATTC-3') and probe (5'-CCTTTGGAAGACCTATT GTACCCGGCA-3') were used to quantitatively determine the copy number of ASFV genomic dsDNA. In a 20 μL reaction system, reagents consisted of 10 μL 2 × Premix Ex TaqTM (Probe qPCR) (Takara), forward primer (0.75 μmol/L), reverse primer (0.75 μmol/L), probe (0.6 μmol/L) and DNA/cDNA templates. All the TaqMan qPCR assays were performed using a CFX96 Real-Time System (Bio-Rad) following the procedures: pre-denaturation at 95 ℃ for 5 min; 40 cycles of denaturation at 95 ℃ for 15 s and annealing at 60 ℃ for 30 s with FAM fluorescent signal detected every minute.
-
This test strip consisted of an absorbent pad, nitrocellulose membrane, colloidal gold pad, sample pad and PVC plastic board. The colloidal gold pad was sprayed with colloidal gold-conjugated anti-FAM mouse IgG antibody (Abcam) at 3.8 μL/cm and the pad was cut into 0.9 cm wide strip. T line (test line) on nitrocellulose membrane was sprayed with 1 mg/mL streptavidin (Solarbio) at 1 μL/cm. C line (control line) on nitrocellulose membrane was sprayed with 0.12 mg/mL protein A (Solarbio) at 1 μL/cm. The nitrocellulose membrane, absorbent pad, colloidal gold pad, sample pad and PVC plastic board were assembled into complete test strips for reporter RNA detection. The intensity of lines was measured by an HR8000 immunoquantitative detector (Wuhan Glary Bio-Tec Co., Ltd, Wuhan, China).
-
Software GraphPad Prism 8 (GraphPad software, Inc., CA) was used for data analysis. Unpaired two-tailed student t test was used to analyze statistical significance. The statistically significant differences were presented at the levels of * P < 0.05, ** P < 0.01, *** P < 0.001 and **** P < 0.0001.
Vector, Viruses and Samples
Preparation of crRNA Targeting Conserved Fragment of ASFV P72 gene
Preparation of CRISPR-lwCas13a Protein
Collateral Detection Combining RPA with CRISPRlwCas13a
Validation of RNA Reporter Cleavage by Electrophoresis
Combination of HUDSON with RPA-CRISPR
TaqMan qPCR Assay
Preparation of Lateral Flow Strip for Detecting Reporter RNA
Statistical Analysis
-
RPA-CRISPR is an in vitro nucleic acid detection platform based on recombinase polymerase amplification and CRISPR-mediated cleavage of the FAM-BHQ RNA reporter (Fig. 1A). The RNA reporter (5'-FAM-UUUUU) and its cleavage product were validated by electrophoresis (Fig. 1B). To specifically detect the viral DNA of ASFV, three crRNAs were validated and applied in RPA-CRISPR (Fig. 1C). The twofold gradient dilutions of crRNA and CRISPR-lwCas13a protein were used for phalanx titration test to determine the optimal concentrations. In this study, 1.626 ng/μL of CRISPR-lwCas13a protein and 2.63 ng/μL of crRNA were determined to be the optimal concentrations for development of RPA-CRISPR detection of ASFV (Fig. 1D, 1E).

Figure 1. Schematic diagram and system optimization of RPA-CRISPR. A Schematic diagram in the whole process of RPA-CRISPR detection. B Analysis of RNA reporter cleavage by acrylamide denaturing electrophoresis. C Validation of crRNAs. D Two-fold gradient dilution of CRISPR-lwCas13a protein was used for determining the optimal concentrations. E Two-fold gradient dilution of crRNA was used for determining the optimal concentrations.
-
RPA-CRISPR is a specific nucleic acid detection method with attomolar sensitivity. To determine the detection limit, tenfold serial dilutions of ASFV P72 plasmid and viral genomic DNA were used for sensitivity assay. P72 plasmid was diluted in sterile water to determine the methodological sensitivity. HUDSON-treated blood suspension containing ASFV genomic DNA was used for sensitivity test. The results indicated that the detection limit was as low as a single copy/μL (100 copy/μL) no matter when RPACRISPR targeted P72 plasmid in water solutions (Fig. 2A) or genomic DNA in blood suspension (Fig. 2B).
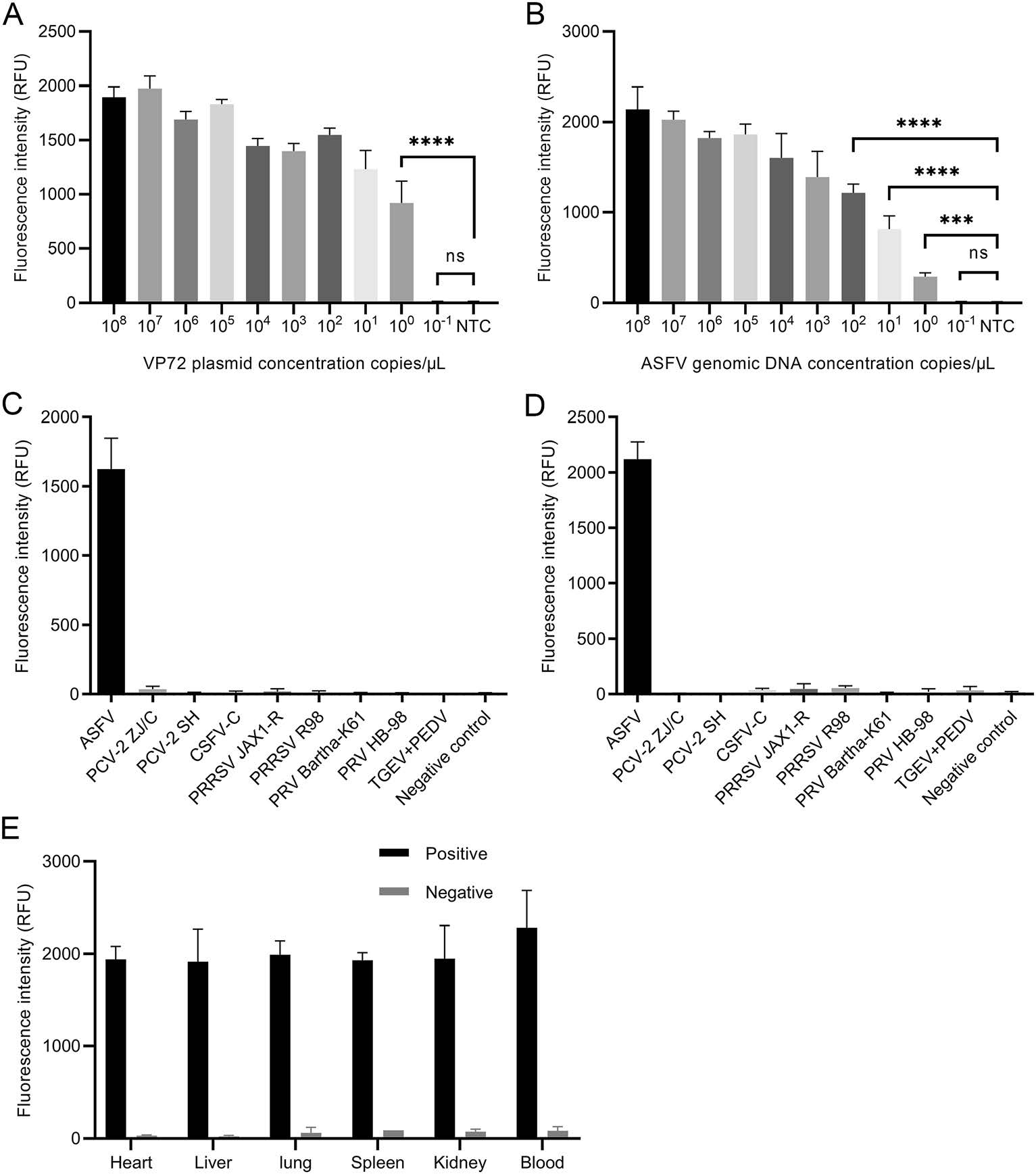
Figure 2. Validation of sensitivity and specificity of qPCR-based RPACRISPR. A tenfold serial dilutions of ASFV VP72 plasmid in ddH2O were used for determining the sensitivity of RPA-CRISPR. B tenfold serial dilutions of ASFV genomic DNA in blood suspension were used for determining the sensitivity of RPA-CRISPR. ASFV viral DNA and control viral DNA or RNA (PCV-2 ZJ/C, PCV-2 SH, CSFV-C, PRRSV JAX1-R, PRRSV R98, PRV Bartha-K61, PRV HB- 98, TGEV + PEDV) were dissolved in sterile water C and blood suspension collected from the SPF pigs D to determine specificity of RPA-CRISPR, respectively. The blood suspension collected from the SPF pigs was used as the negative control. E ASFV viral DNA was separately added in different tissue suspensions to be analyzed by RPA-CRISPR. Tissue suspension from the SPF pigs was included as a negative control.
-
Viral genomic DNA or RNA was extracted from ASFV positive blood samples and commercial vaccines of other porcine viruses (PCV-2 ZJ/C, PCV-2 SH, CSFV-C, PRRSV JAX1-R, PRRSV R98, PRV Bartha-K61, PRV HB-98, TGEV and PEDV) and was dissolved in either sterile water or blood suspension collected from specific-pathogen-free (SPF) pigs. ASFV genomic DNA and other control viral DNA or RNA were used for determining the specificity of RPA-CRISPR. The results indicated that RPA-CRISPR accurately distinguished viral DNA of ASFV from control viral DNA or RNA with no cross-reactivity in either water or blood suspension (Fig. 2C, 2D). ASFV viral DNA (50 ng) was separately added into 1 mL of different tissue suspensions (heart, liver, lung, spleen, kidney and blood) to validate whether cellular components from different tissues affect RPA-CRISPR reaction. Each type of tissue suspension containing no ASFV viral DNA was included as a negative control. The results indicated that RPA-CRISPR could accurately distinguish the ASFV viral DNA from negative control in different tissue suspensions (Fig. 2E).
-
Since the specificity of RPA-CRISPR depends on the number of mismatches on crRNA target region, sequences of ASFV different genotype strains were compared to identify the mismatches. The results indicated that no mismatch could be found at the target regions of all crRNAs on ASFV P72 gene (genotype Ⅰ, Ⅱ, Ⅲ, Ⅳ, Ⅴ and ⅩⅩ) (Table 1). The genotype Ⅸ and Ⅹ could not be detected by crRNA-1, while genotype Ⅶ, Ⅸ and Ⅹ could not be identified by crRNA-3. crRNA-2 could be used for detecting all the genotypes analyzed in this study (Table 1).
Isolate name GenBank no Country Genotype Mismatches cr1a cr2b cr3c E75 FN557520.1 Spain Ⅰ 0 0 0 Georgia 2007/1 NC_044959.1 Georgia Ⅱ 0 0 0 Wuhan 2019–1 MN393476.1 China Ⅱ 0 0 0 Krasnodar 2012 KJ195685.1 Russia Ⅱ 0 0 0 Pig/HLJ/2018 MK333180.1 China Ⅱ 0 0 0 Warmbaths AY261365.1 South Africa Ⅰ/Ⅲ 0 0 0 Warthog AY261366.1 Namibia Ⅳ 0 0 0 Tengani 62 AY261364.1 Malawi Ⅴ 0 0 0 Mkuzi 1979 AY261362.1 South Africa Ⅰ/Ⅶ 0 0 2 Ken06.Bus NC_044946.1 Kenya Ⅸ 2 0 2 Ken05/Tk1 KM111294.1 Kenya Ⅹ 2 0 2 Pretoriuskop/96/4 AY261363.1 South Africa ⅩⅩ/Ⅰ 0 0 0 a, b, c crRNA-1, crRNA-2 and crRNA-3, respectively Table 1. Analysis of mismatches on crRNA targeted regions of P72 genes from different genotype strains.
-
To compare RPA-CRISPR with the traditional PCR method, a TaqMan qPCR-based method was established according to the previous studies (King et al. 2003). The tenfold serial dilution of ASFV P72 plasmid was used for constructing standard curve and determining the sensitivity of TaqMan qPCR. A standard curve with R2 of 0.994 was constructed for quantitative detection of viral DNA copy numbers (Fig. 3A). The detection limit of qPCR was 101 copies/μL when ASFV P72 plasmid was used as PCR template (Fig. 3B). In specificity test, viral genomic DNA and RNA of ASFV and other porcine viruses were used for qPCR detection. The results indicated that qPCR assay could accurately distinguish ASFV viral DNA from control viral DNA or RNA with no cross-reactivity (Fig. 3C).

Figure 3. Sensitivity and specificity validation of qPCR. A tenfold serial dilutions of ASFV VP72 plasmid were used for constructing the standard curve of qPCR. B tenfold serial dilutions of ASFV VP72 plasmid were used for sensitivity validation of qPCR. C ASFV viral DNA and control viral DNA or RNA (PCV-2 ZJ/C, PCV-2 SH, CSFV-C, PRRSV JAX1-R, PRRSV R98, PRV Bartha-K61, PRV HB-98, TGEV + PEDV) were dissolved in sterile water for determining specificity of qPCR.
To validate the repeatability, variation coefficients of intra-assay and inter-assay were analyzed by using the Ct value obtained from qPCR assay of 10 folds serially diluted plasmid (5 × 107 copies/μL – 5 × 105 copies/μL). The results indicated that the intra-assay (Table 2) and interassay (Table 3) variation coefficients were less than 3%. Therefore, this qPCR assay exhibited a robust repeatability both in intra-assay and inter-assay.
Plasmid concentration (copies/μL) Ct value sample 1 Ct value sample 2 Ct value sample 3 Standard deviation(SD) Variation coefficient(CV) 5 × 107 12.11 12.20 12.21 0.055 0.45% 5 × 106 15.36 15.40 15.46 0.050 0.32% 5 × 105 18.57 18.57 18.65 0.046 0.24% Table 2. Intra-assay variation coefficient of qPCR.
Plasmid concentration (copies/μL) Ct value sample 1 Ct value sample 2 Ct value sample 3 Standard deviation(SD) Variation coefficient(CV) 5 × 107 12.09 12.47 12.75 0.331 2.66% 5 × 106 15.36 15.58 16.26 0.469 2.98% 5 × 105 18.73 19.21 19.26 0.296 1.53% Table 3. Inter-assay variation coefficient of qPCR.
-
In this lateral flow strip-based RPA-CRISPR assay, RNA reporter was labeled with a 5' FAM and a 3' biotin for detecting the collateral cleavage effect. The detection product of RPA-CRISPR was tenfold diluted and then was added into sample pad on the strip for determining the detection result (Fig. 4A). The concentration of the streptavidin was previously reduced to make both Test (T) line and Control (C) line appear as two consistent bands for negative control when preparing strips in the method section. Under this condition, the result judgment standard of this strip was depended on the degree of elimination on T line. Briefly, a high-intensity band was observed at C line, but not at T line of the strip due to the cleavage of reporter RNA induced by ASFV positive samples. In contrast, in ASFV negative samples, high-intensity bands were observed at both C and T lines, since the colloidal gold conjugated anti-FAM antibody was immobilized on T line by not-cleaved reporter RNA. The intensity ratio of T to C line was calculated to reveal significant difference between ASFV weakly positive specimen and negative control (Fig. 4B).
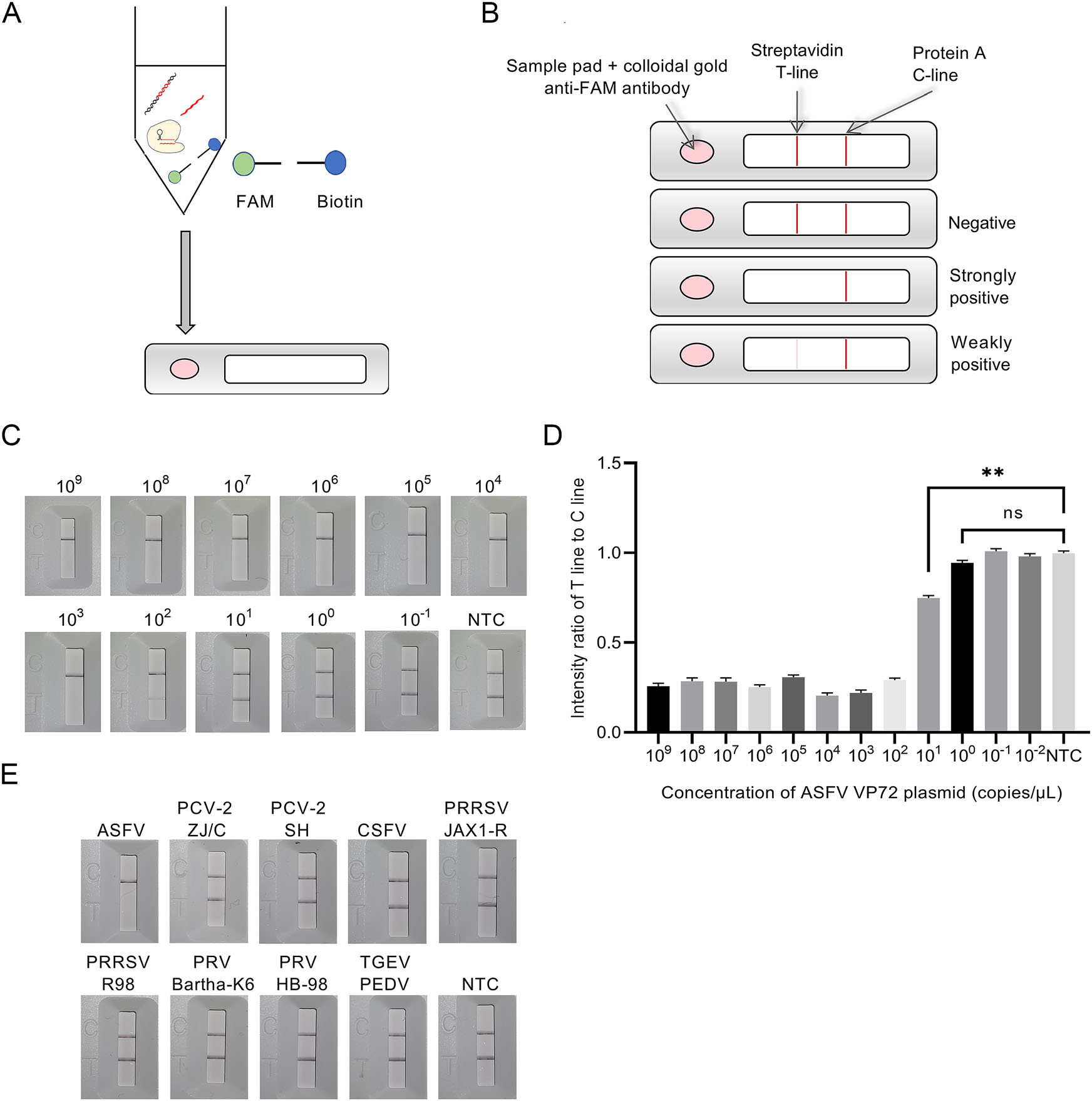
Figure 4. Schematic diagram of detection process, sensitivity and specificity of lateral flow strip-based RPA-CRISPR. A Detection process of lateral flow strip-based RPA-CRISPR. B Detection method of lateral flow strip-based RPA-CRISPR. C Sensitivity of lateral flow strip-based RPA-CRISPR with visual observation. D Intensity ratio of T line and C line in sensitivity test of flow strip-based RPA-CRISPR. E Specificity of lateral flow strip-based RPA-CRISPR.
-
Since the collateral cleavage effect was determined by using a lateral flow strip instead of a qPCR system, sensitivity and specificity of this lateral flow strip-based RPACRISPR assay needed to be further evaluated. Lateral flow strip-based RPA-CRISPR could detect as low as 102 copies/μL with visual observation (Fig. 4C). The T/C line intensity ratio obtained by an HR8000 immuno-quantitative detector indicated that the plasmid at concentration of 101 copies/μL (Fig. 4D) was ASFV weakly positive. Specificity test results revealed that the lateral flow stripbased RPA-CRISPR could accurately distinguish ASFV viral DNA from control viral DNA or RNA with no crossreactivity (Fig. 4E).
-
In order to validate the reliability of clinical sample detection, the results of qPCR-based RPA-CRISPR, lateral flow strip-based RPA-CRISPR and qPCR were compared. Total 27 ASFV positive specimens and 25 ASFV negative specimens were applied in all 3 different assays mentioned above. The results of qPCR-based RPA-CRISPR and lateral flow strip-based RPA-CRISPR were compared with those of qPCR. The coincidence rates of ASFV positive samples and ASFV negative samples were calculated. The results demonstrated that the detection accuracy of qPCR and qPCR-based RPA-CRISPR exhibited a coincidence rate as high as 100% for both all ASFV positive and negative samples (Fig. 5A). The lateral flow strip-based RPA-CRISPR assay displayed 8 false-negative results obtained from the 27 detected ASFV positive samples with the coincidence rate of 70.3%, compared with qPCR (Fig. 5A). Notably, the results of the 25 ASFV negative samples by using three methods were completely identical with coincidence rate of 100% (Fig. 5B).
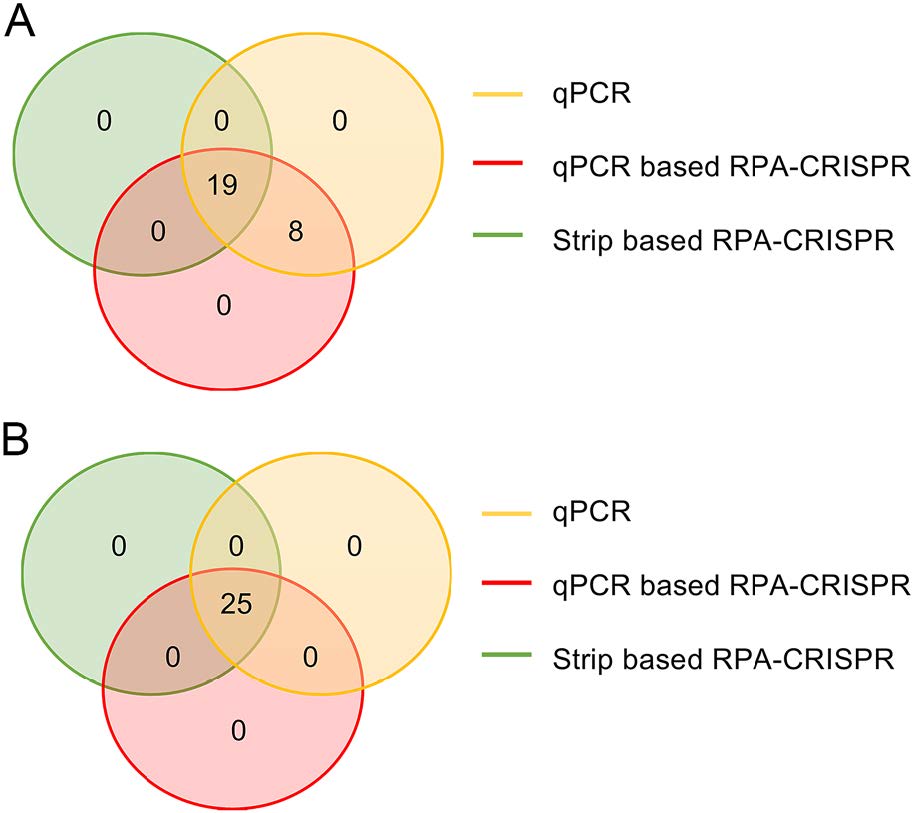
Figure 5. Coincidence rate of ASFV clinical sample assay results by using three different detection methods. A Coincidence rate of 27 ASFV positive clinical sample assay results. B Coincidence rate of 25 ASFV negative clinical sample assay results.
Optimization of crRNA and CRISPR-lwCas13a in the Reaction System
Validation of Detection Limits of ASFV P72 Plasmid and ASFV Genomic DNA by qPCR-based RPA-CRISPR
Specificity Determination of qPCR-Based RPACRISPR
Sequence Analysis of crRNA Target Region on ASFV P72 Gene
Sensitivity, Specificity and Repeatability Validation of TaqMan qPCR
Construction of the Lateral Flow Strip and Its Combination with RPA-CRISPR
Sensitivity and Specificity of Lateral Flow StripBased RPA-CRISPR
Clinical Sample Detection by Three Different Assays
-
Since the first outbreak of ASF in China on August 3, 2018, immediate emergency response and other control strategies have been initiated and applied to reduce disease spreading (Ge et al. 2018). However, continual outbreaks are still ongoing in different regions due to complex factors (Kim et al. 2019; Zhao et al. 2019). Currently, ASF control strategies largely depend on rapid detection and slaughter of infected pigs. It is urgent to develop a quick detection method for diagnosing the suspected swine herds in pig farms or enterprises. In this study, a novel RPA-CRISPR assay was developed as a suitable method with high sensitivity, rapidity and specificity.
It was reported that blood samples had a high level of viral load in ASFV infected pigs (Olesen et al. 2017; Dixon et al. 2019). The HUDSON treatment method was used for inactivating the protease and nuclease to protect the ASFV genomic DNA (Gootenberg et al. 2017). Afterwards, ASFV genomic DNA was directly amplified by RPA to avoid additional time for DNA extraction (Piepenburg et al. 2006). Subsequently, the amplicon of RPA was further converted into target ssRNA through transcription for activating CRISPR-mediated collateral cleavage of reporter RNA with whole detection process lasting no more than 1.5 h. Otherwise, it would take at least one hour in DNA extraction by using either phenol–chloroform method or commercial DNA extracting kits. RPA-CRISPR displayed a great superiority in detection speed, therefore it had great potential to be applied for rapid diagnosis.
There were two methods for detection of the cleaved reporter RNA, namely, qPCR and lateral flow strip, which were used for laboratorial routine diagnosis and potential application for field diagnosis, respectively. The detection results were presented by the difference in FAM fluorescent intensity between final cycle and initial cycle. As indicated in our study, the detection limit of qPCR-based RPA-CRISPR assay was as low as a single copy (100 copy/μL). RPA-CRISPR exhibited the same or even higher sensitivity than the traditional qPCR method recommended in OIE terrestrial manual (Haines et al. 2013; King et al. 2003). In addition, RPA-CRISPR was more sensitive than those ASFV detection methods that reported in other studies, such as a OIE-validated PCR assay, a novel PCR assay, a loop-mediated isothermal amplification (LAMP) assay and an RPA-based assay (Luo et al. 2017; Wang D et al. 2019, 2017; Fernandez-Pinero et al. 2013). Our RPA-CRISPR also exhibited a same level of sensitivity compared with that of CRISPR methods for detecting Influenza A virus (H7N9), Ebola virus and Mycobacterium tuberculosis (Liu et al. 2019; Qin et al. 2019; Ai et al. 2019). The visual observation results indicated that detection sensitivity of lateral flow strip-based RPA-CRISPR was 102 copies/μL. Previous study also reported a CRISPR visual detection method for PRRSV with a same level of sensitivity (Chang et al. 2019). To overcome the limitations of visual inspection, a portable appliance (HR8000 immuno-quantitative detector) was applied instead of visual detection and was found to have the same level of sensitivity (101 copies/μL) with TaqMan qPCR assay. However, in comparison with qPCR-based RPA-CRISPR, strip-based RPA-CRISPR exhibited a relatively lower sensitivity, but not significant, which might be attributed to the fact that the weak collateral cleavage effect induced by a single copy of DNA was not enough to exhibit a significant difference between the examined samples and negative control on the lateral flow strip. Therefore, it was suggested that a cascade amplification effect should be considered for enlarging the weak collateral cleavage effect induced by a single copy of DNA when using the method of RPA-CRISPR in the future study.
The lateral flow strip is a useful tool for rapid detection (Quesada-Gonzalez et al. 2015; Ngom et al. 2010). A lateral flow strip was reported to be constructed by using a FAM-biotin RNA reporter and the streptavidin-and-protein-A-coated strip (Myhrvold et al. 2018; Gootenberg et al. 2018). Previous study also reported that when negative samples was detected, the capture of the colloidal gold conjugated anti-FAM antibody by the protein A line could be avoided by binding the biotin molecules to immobilize all of the reporter RNAs on the streptavidin line (Gootenberg et al. 2018). To achieve this purpose, high concentration of streptavidin was required to immobilize the abundant reporter on the streptavidin line, resulting in high cost and technical difficulty in production process. If reporter RNA is a little degraded by RNase in the air or long-time storage, a weak intensity on protein A line will be observed, which may lead to a false positive result by the judgement standard in previous study. In this study, a reduced concentration of streptavidin was used at T line, leading to less expenditure on the strip. When reporter RNA is a little degraded, intensity reduce on T line will not cause a significant difference on T/C ratio to avoid the false positive result according to the judgement standard in this study. Therefore, the advantage of lateral flow strip lay in less expenditure and less false positive result in this study.
To verify the availability of RPA-CRISPR in ASFV clinical diagnosis, 27 ASFV positive samples and 25 negative samples were analyzed to calculate the coincidence rate between RPA-CRISPR and qPCR. Due to sensitivity limitation, no significant difference in intensity was observed between T-line and C-line in the assay of lateral flow strip-based RPA-CRISPR, when small number of reporter RNAs were cleaved. The lateral flow strip-based RPA-CRISPR assay correctly detected 70.3% of all the 27 ASFV positive DNA samples. On one hand the lateral flow strip-based RPA-CRISPR may have the potential for preliminary screening of ASFV infection at porcine farms or enterprises. On the other hand, the qPCR-based RPACRISPR is available to the laboratory diagnosis of ASFV clinical cases with the coincidence rate of 100% for both ASFV positive and negative samples.
In summary, this study demonstrates that RPA-CRISPR is a rapid, robust and sensitive method for ASFV diagnosis and this method has great potential in future applications to swine industry and food security.
-
This research was supported by the National Natural Science Foundation of China (31522057 and 31872451 to LZ; 31720103917 and 31872452 to ZF).
-
MR, LZ, ZF and DB designed the experiments. MR, MZ and HM carried out the experiments. MR, MZ and LZ analyzed the data. MR and LZ wrote the paper. FP provided the samples. MR, HH and LZ checked and finalized the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This study was performed in accordance with recommendations in the care and use of laboratory animals of the Ministry of Science and Technology of China and was approved by the Scientific Ethics Committee of Huazhong Agricultural University (permit number HZAUSW-2019–007 approved on May 16, 2019).

 DownLoad:
DownLoad: