












HTML
-
Bacteriophages are viruses that attack bacteria and kill them through the lytic replication cycle. Many studies have reported that phages are more specific to bacteria than antibiotics are; thus, phage therapy has many potential applications in human medicine, with the advantage of having few side effects (Keen, 2012). Investigating the mechanisms of bacteria-killing phages will therefore aid in the development of antibacterial drugs.
Hydrolases encoded by phages play a key role in the interaction between phages and host bacteria. These enzymes act on the bacterial cell wall to kill the host bacteria and then release progeny phages (Nielsen et al., 1999). Thus, correctly identifying the hydrolases encoded by phages can provide important clues for not only studying the lytic mechanism of the phage-bacteria system but also discovering potential antibacterial drugs. With the accumulation of proteomics data, various machine-learning methods have been applied to predict functional phage proteins. Sequritan et al. designed an artificial neural network (ANN)-based method to predict viral structural proteins using amino acid frequency (Seguritan et al., 2012). Recently, a special type of structural protein, phage virion protein, was identified using primary sequence information (Ding et al., 2014; Feng et al., 2013).
However, to our knowledge, no computational method has been developed to predict phage hydrolases. Thus, the aim of this letter is to describe a powerful model for identifying phage hydrolases. We started by discrimi-nating phage enzymes from phage non-enzymes. Once a phage protein is recognized as phage enzyme, the model will determine whether the predicted enzyme is phage hydrolase.
First, we collected phage proteins from the Universal Protein Resource (UniProt) (UniProt, 2015). To improve the quality of the data, we only chose phage proteins that have been annotated in Swiss-Prot. Subsequently, we excluded proteins whose sequences contained illegal chara-cters such as "B", "X", and "Z". Furthermore, the program CD-HIT (Fu et al., 2012) was used to eliminate similar sequences with a cutoff threshold of 30%. Finally, the benchmark dataset contained 124 phage enzymes and 131 phage non-enzymes. The 124 phage enzymes were divided into 69 hydrolases and 55 non-hydrolases.
Second, we used the g-gap dipeptide composition extending from the adjoining dipeptide composition to describe the correlation of the residues in the protein primary sequence (Lin et al., 2013). Thus, a given phage protein P can be formulated by a 400-dimension vector. Based on the hypothesis that if the sample variance of a feature between groups is larger than the sample variance within groups, then the feature is suitable for classification, we used analysis of variance (ANOVA) (F-scores) to describe the contribution of each feature (Lin et al., 2013).
Third, we used a support vector machine (SVM) to perform the classification. The free software package LibSVM was used to implement the SVM. The radial basis function (RBF) was chosen as the kernel function because it is more suitable for nonlinear classification than other kernel functions (Tang et al., 2016). To obtain the best performance, the grid search approach was applied to optimize the regularization parameter C and the kernel width parameter γ. The performance of our models was quantitatively evaluated using jackknife cross-validation tests (Guo et al., 2014; Lin et al., 2008; Lin et al., 2014; Liu et al., 2015) with the use of three indexes (Lin et al., 2013; Zhu et al., 2015; Zou et al., 2013): sensitivity (Sn), specificity (Sp), and overall accuracy (OA).
Following the above steps, we first needed to deter-mine whether a phage protein was an enzyme. For this step, we varied the interval residue parameter g from 0 to 9. Our proposed feature selection technique described in this letter was used to exclude noise and redundant information. An arbitrary g-gap dipeptide composition has 400 features, and a total of 400 F-scores were calculated for the 400 features. Subsequently, the 400 features were ranked according to their F-scores. The incremental feature selection (IFS) process was used to determine the optimal number of features according to the following steps. First, the feature with the highest F-score was selected as the SVM input. The OA was then calculated to evaluate the performance of this feature. Second, the feature with the second highest F-score was combined with the first feature to form a new feature subset. The OA was also used to estimate the performance of the new feature subset using the SVM. This process was repeated until 400 OAs were calculated. The best feature subset is defined as the subset that can produce the highest OA. By setting dimension of the feature subset (the number of features) as the abscissa and the OA as the ordinate, we plotted the 10 curves shown in Figure 1A. As shown in this figure, the highest OA of 84.3% can be achieved by 59 2-gap dipeptides, which are regarded as the optimal feature subset. Thus, the first model was constructed using these features, with Sn and Sp values of 87.1% and 81.7%, respectively.
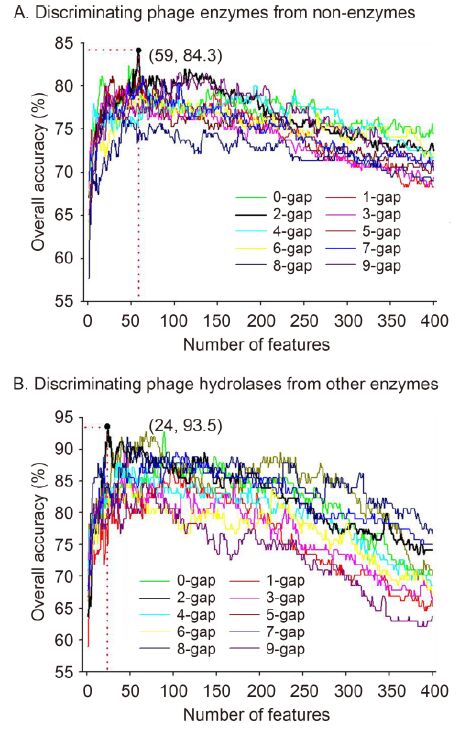
Figure 1. A plot showing the incremental feature selection (IFS) procedure for (A) discriminating phage enzymes from non-enzymes and (B) discriminating phage hydrolases from other enzymes.
Once a new sequenced phage protein is discriminated as a phage enzyme, the second step is to determine whether the phage enzyme is a hydrolase. As in the first step, the parameter g was varied from 0 to 9. Moreover, the same feature selection process described in the above paragraph was used to identify the best feature subset that can produce the highest OA. Accordingly, we examined the predictive performance of 4, 000(400 × 10) feature subsets and obtained the 10 curves shown in Figure 1B. The results show that the highest OA of 93.5% can be achieved by the optimized feature subset including 24 5-gap dipeptides. In addition, 92.8% of phage hydrolases and 94.5% of other phage enzymes were correctly identified in jackknife cross-validation tests. These results indicate that the genetic information for phage hydrolases is mainly contained in higher-order correlations, and these should be further investigated to determine their biological meaning.
Based on the above model, we built a user-friendly webserver called PHYpred that can be used by the vast majority of scholars to efficiently and easily study phage enzymes and hydrolases without having to learn complicated mathematics or programs. The web interface used to browse and submit entries is coded in PHP. The server can be freely accessed at http://lin.uestc.edu.cn/server/PHYPred. A guide on how to use the tool to obtain the desired results is provided in the webserver.
The correlation of nucleotides or residues is the main carrier of genetic information. Therefore, we used g-gap dipeptide compositions as features for prediction. However, the performances of models based on such fundamental information are far from satisfactory. To improve the accuracies and identify the real correlations hidden in protein sequences, a feature selection technique was applied to select optimal features. The results demonstrate that the technique can pick out informative features, dramatically improve the predictive perfor-mance, and enhance the generalizati on abilities of the pro-posed models. Using the correlation information and feature selection technique, our models produced promising results for predicting phage enzymes and hydrolases.
In summary, a new tool called PHYPred was established for the accurate prediction of potential novel phage enzymes and hydrolases. In PHYPred, a high-quality benchmark dataset was constructed by setting a series of standards, which can guarantee the reliability of the tool. Thus, the dataset has the potential to become a standard dataset for user in the development of computational methods for the prediction of phage enzymes and hydrolases. Moreover, a feature selection technique was successfully applied to improve the performance. Our results indicated that the proposed model can predict phage enzymes and hydrolases at a high discriminative accuracy. This method can also be used in other fields such as bioinformatics and computational biology.
-
This work was supported by the National Nature Scientific Foun-dation of China (No. 61301260), the Applied Basic Research Program of Sichuan Province (No. 2015JY0100 and LZ-LY-45), the Scientific Research Foundation of the Education Department of Sichuan Province (11ZB122), the Nature Scientific Foundation of Hebei Province (No. C2013209105), the Fundamental Research Funds for the Central Universities of China (No. ZYGX2015J144, ZYGX2015Z006), and Program for the Top Young Innovative Talents of Higher Learning Institutions of Hebei Province (No. BJ2014028). The authors declare no conflict of interest. This arti-cle does not contain any studies with human or animal subjects performed by any of the authors.


 DownLoad:
DownLoad: