














HTML
-
Zika virus (ZIKV) is an emerging pathogen, transmitted by Aedes species, that belongs to the genus Flavivirus of the family Flaviviridae. ZIKV-infected patients usually have symptoms characterized by fever, headache, rash, myalgia, arthralgia, and conjunctivitis, which are similar to the clinical symptoms caused by Dengue virus (DENV) or Chikungunya virus (CHIKV) infection. Thus, ZIKV could be easily misdiagnosed as Dengue (DEN) or Chikungunya (CHIK) fever (Haddow et al., 2012). A body of evidence has shown that ZIKV infection is associated with the neurological disorders microcephaly (Mlakar et al., 2016) and Guillainâ€"Barré syndrome (Oehler et al., 2014). Further, not only has the number of ZIKV cases increased in recent years, but the disease has spread to new regions that had no previous record of Zika disease. Thus, Zika disease has become a serious public health problem. The worldwide dispersal and transmission of ZIKV should be of great concern to researchers, and their study will contribute to the prevention and control of outbreaks and epidemics of Zika disease.
ZIKV first emerged in Africa. It was first isolated in 1947 from a sentinel monkey in Uganda (Dick et al., 1952). The first ZIKV infection in human was identified in Uganda and Tanzania in 1952 (Smithburn, 1952). ZIKV was detected in humans, Aedes species and animals in African countries in the following decades (Macnamara, 1954; Simpson, 1964; Faye et al., 2014). Then, ZIKV spread to Southern Asia. It was isolated from Aedes aegypti in Malaysia in 1966 (Marchette et al., 1969). The wide distribution of ZIKV in Indonesia, Malaysia, and Pakistan was further revealed by serological investigations (Kindhauser et al., 2016). Subsequently, ZIKV spread to countries in Oceania and the Americas. The first large outbreak of Zika disease in humans occurred on the Pacific island of Yap in the Federated States of Micronesia in 2007 (Lanciotti et al., 2008; Duffy et al., 2009). ZIKV was then identified from Suriname (Enfissi et al., 2016b) in South America, and on other Pacific Islands including French Polynesia (Berthet et al., 2014; Cao-Lormeau et al., 2014; Hancock et al., 2014), Easter Island of Chile (Tognarelli et al., 2015), the Cook Islands (Roth et al., 2014), and New Caledonia (Roth et al., 2014; Dupont-Rouzeyrol et al., 2015). Imported cases were also identified in countries of the Americas and Europe (Foy et al., 2011; Waehre et al., 2014; Zammarchi et al., 2015; Kindhauser et al., 2016). Until March 2016, a total of 10 imported infectious cases had been diagnosed in China. A brief review of ZIKV's history found that ZIKV migration seems to have followed the same path as CHIKV and DENV, which were first described in Africa and then spread to Asia before becoming distributed worldwide (Musso et al., 2015). Although the future is unpredictable, ZIKV has the potential to spread globally through the increasing international movement of humans, goods, and animals for travel and business, and could represent a major threat to human health. Investigating the migration pathways of ZIKV can help to shed light on the origins of ZIKV epidemics.
Two major genetic lineages of ZIKV strains associated with its geographic distribution, namely the African and Asian lineages, have been characterized and limited ZIKV nucleic acid sequences are available (Haddow et al., 2012; Faye et al., 2014). The strains identified from recent epidemics in the Americas belonged to the Asian lineage (Enfissi et al., 2016b), suggesting that strains from Asia and the Americas are closely related. However, as only limited nucleic acid sequences of ZIKV strains are available in GenBank for phylogenetic analysis and other bio-informatics analyses (Haddow et al., 2012; Faye et al., 2014), knowledge of the genetic relationships of ZIKV strains and the geographic origins of ZIKV epidemics is limited. This study focuses on the phylogenetic relationships among ZIKV strains, the spatial and temporal distributions of ZIKV lineages, and the potential recombination and migration of ZIKV among different countries and territories. The results reveal the geographic origins and migration patterns of ZIKV epidemics, which will be further discussed.
-
ZIKV sequences deposited in GenBank until February 4, 2016 were used for phylogenetic analysis. Sequences were aligned by CLUSTAL W (MEGA 5.0). Phylogenetic trees were constructed for the full-length genome, the envelope protein (E) coding region, and the non-structural protein 5 (NS5) coding region by MEGA 5.0 using both the neighbor-joining (NJ) and maximum-likelihood (ML) methods, and tested by the bootstrap method with 1000 replicates. The sequence of Spondweni virus (GenBank accession number: DQ859064) was included as the outgroup control.
-
Recombination events were detected by the RDP software package using seven recombination detection methods (Martin et al., 2010) based on the concatenate sequences of E protein coding region and NS5 protein coding region. Recombination events that were significant by at least two methods (P < 0.05) and had an RDP recombination consensus score (RDPRCS) of more than 0.60 were considered to be confirmed events. Recombination events that were significant by at least two methods (P < 0.05) and had an RDPRCS score between 0.4â€"0.6 were considered to be possible events. Events that did not meet these criteria were rejected.
-
The ZIKV spread pathway was analyzed using the MIGRAPHYLA package (Wallace et al., 2007) based on the NS5 tree. The reliability of each migration event was evaluated using a Monte Carlo simulation process by rand omizing the location of the leaf nodes while retaining the tree topologies.
Sequence alignment and construction of phylogenetic trees
Recombination detection
Virus migration pathway analysis
-
In total, 23 full-length genomic sequences of ZIKV were included in the genomic tree, representing two lineages related to the geographic distribution of ZIKV as described previously; namely, the African lineage and the Asian lineage (Figure 1) (Haddow et al., 2012; Enfissi et al., 2016b). The Asian lineage includes ZIKV strains derived mainly from countries in Asia and the Americas, and the African lineage is composed only of ZIKV strains derived from Africa (Figure 1). Hence, ZIKV evolution is associated with its geographic distribution, although the African and Asian lineages evolved separately.

Figure 1. A phylogenetic tree of ZIKV strains based on their full-length genome sequences. Two lineages were identified and designated as the African lineage and the Asian lineage. The strains that were assigned to different phylogenetic positions among the full-length genomic tree, E tree, and NS5 tree (Figure 2) are labeled by black solid circles (●).
Robust E and NS5 trees were constructed by the NJ and ML methods, based on the E and NS5 coding regions available in GenBank (Figure 2). The NJ and ML trees presented identical topological structures. Besides the two main lineages, one more lineage was found in both the E tree (Figure 2A) and the NS5 tree (Figure 2B). This extra lineage was not found in the full-length genome tree, probably because few full-length genomic sequences of ZIKV are available in GenBank besides these new lineage sequences. According to the geographic distributions of the strains clustering into each lineage, the three lineages were designated as the Asian/American lineage, African lineage 1, and African lineage 2 (Figure 2). African lineage 2 evolved separately and diverged early from the other two lineages (Figure 2). In the E tree, it was composed of strains from Senegal in 1998 and 2001 and one strain from Cote d'Ivoire in 1980 (ArA1465) (Figure 2A). In the NS5 tree, the strain from Cote d'Ivoire (ArA1465) clustered in African lineage 1 (Figure 2B). These results indicated that primitive strains might circulate in Senegal and Cote d'Ivoire. Moreover, other strains from Senegal and Cote d'Ivoire were included in African lineage 1 (Figure 2A and B). These results suggested that at least two distinct lineages of ZIKV strains have co-circulated in these two countries.
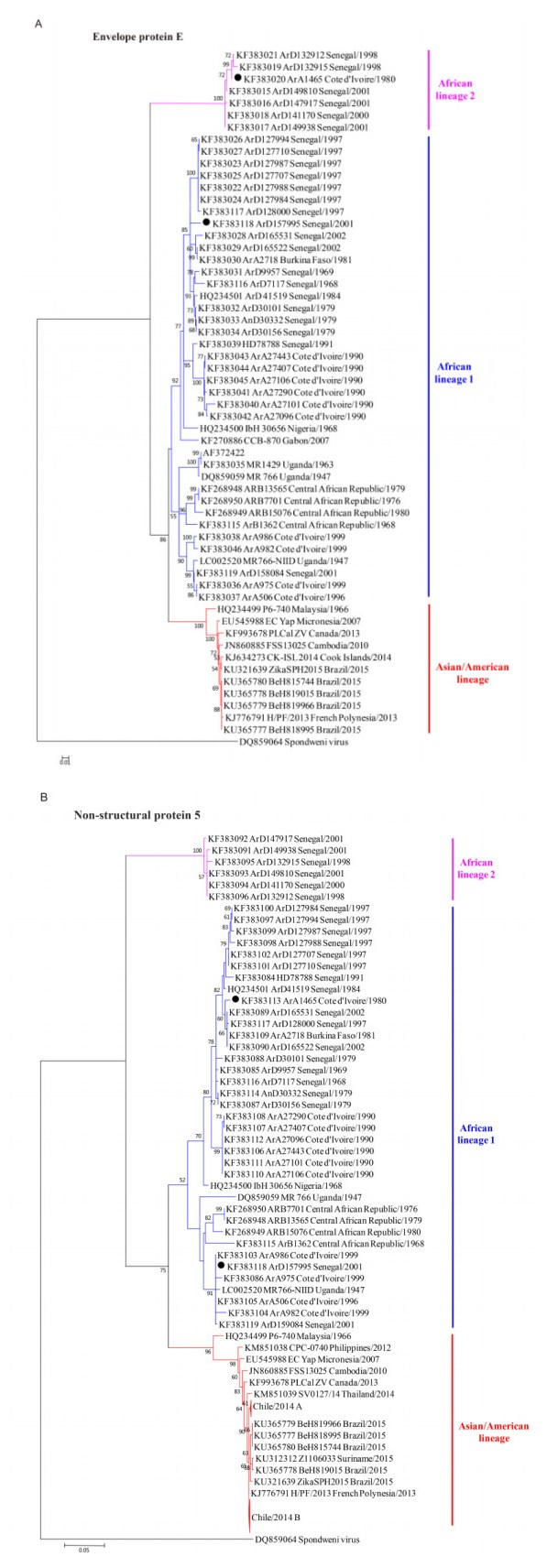
Figure 2. Phylogenetic trees of ZIKV strains based on their envelope protein (E) coding regions (A) and non-structural protein 5 (NS5) coding regions (B). Three lineages were identified in the E and NS5 trees, and were designated as the Asian/American lineage, African lineage 1 and African lineage 2 according to their geographic distributions. The strains present at different phylogenetic positions among the full-length genomic tree (Figure 1), E tree, and NS5 tree are labeled by black solid circles (●). Chile/2014 A and Chile/2014 B represent the compressions of two clusters of strains identified from the Easter Island of Chile.
-
The global ZIKV epidemic appears to have been increasing in terms of the number of infections and spreading to new areas in recent years. Until Feb 2016, 73 countries and territories distributed in Africa, Europe, Asia, the Americas, and Oceania have confirmed individual ZIKV cases and ZIKV epidemics according to data from the CDC and WHO surveillance networks and national public health authorities (Figure 3A, Table 1). Autochthonous outbreaks of ZIKV were confirmed in most countries within the area at risk of DENV epidemics, which also coincided with the areas of CHIKV outbreaks (Figure 3A, indicated in purple color). Imported ZIKA infection cases were confirmed in 24 countries and territories (Figure 3A, indicated in yellow color, and Table 1), since the patients all had a history of travel to ZIKV epidemic areas. ZIKV isolates and strains with available genomic information were identified from 20 countries and territories (Table 1, Figure 3A, indicated in red characters).
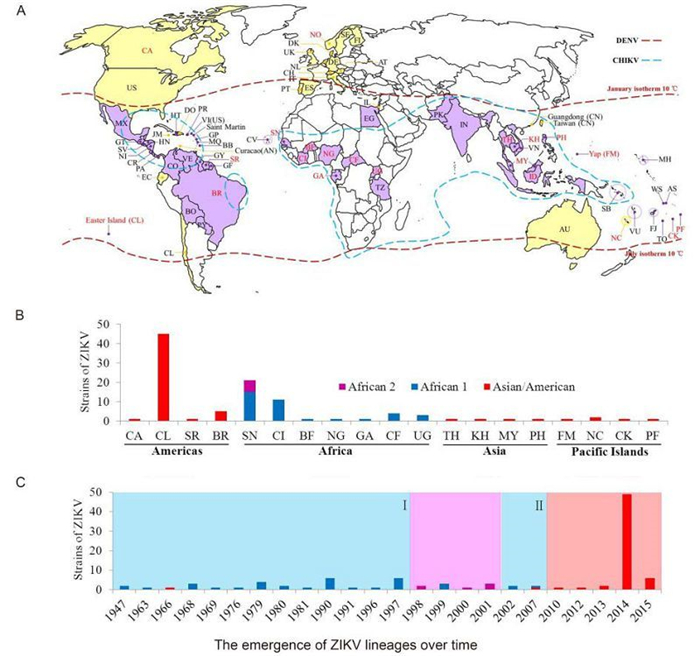
Figure 3. The spatial and temporal distributions of ZIKV lineages worldwide. (A) A world map illustrating the countries and territories that have reported confirmed ZIKV infections. Countries and territories with autochthonous ZIKV outbreaks are indicated in purple color, while those with confirmed imported cases are indicated in yellow color. Countries and territories that have reported ZIKV outbreaks and confirmed cases are labeled with their abbreviated names as shown in Table 1. Countries and territories that could be traced from the annotation of the complete or partial sequences of ZIKV strains deposited in GenBank are labeled in red characters. The areas at risk of DENV infection are delineated by the geographic limits between the northern and southern hemispheres indicated by the dash lines in dark red color. The countries and areas at risks of CHIKV infection are delineated by the dash lines in cyan color. (B) A summary of the numbers of ZIKV strains from different lineages found in different geographic areas. (C) The emergence of ZIKV lineages over time. The periods of time (marked Ⅰ and Ⅱ) of the African lineage 1 epidemics are indicated by a light cyan background color; that of the African lineage 2 epidemics by a lavender background color; and that of the Asian/American lineage epidemics by a vermilion background color. The annotations Asian/American, African 1, and African 2 denote the lineages of the same names.
Table 1. Summary of the countries and territories with a history of ZIKV disease and reported cases
The phylogenetic lineages from different geographic distributions were summarized based on the combined datasets of both E and NS5 sequences (Figure 3B). Viruses of the Asian/American lineage were circulating mainly in American and Asian countries as well as the Pacific Islands, while viruses of African lineages 1 and 2 were circulating in African countries. The prevalence of ZIKV lineages over time was also characterized based on the information of the strain sequences based on the combined datasets (Figure 3C). Outbreaks and epidemics of ZIKV strains of African lineage 1 were identified from 1947 to 1997 as well as in 2002 and 2007 (Figure 3C, light cyan pattern Ⅰ and Ⅱ), whereas only two Asian/American lineage strains was isolated, one from Malaysia in 1966 (Marchette et al., 1969) and the other from Yap Island in the Federated States of Micronesia in 2007 (Lanciotti et al., 2008). Then the African lineage 2 strains from Senegal were identified from 1998 to 2001 (Figure 3C, lavender pattern). Since 2010, ZIKA diseases were reported to be caused by strains of the Asian lineage (Figure 3C, vermilion pattern).
-
Three lineages were identified according to our phylogenetic analysis. However, two strains had different phylog-enetic positions within lineages and clusters among the full-length genomic tree, the E tree, and the NS5 tree (Figure 1, Figure 2A and Figure 2B, black solid circles, and Supplementary Table S1). Strain ArD157995 from Senegal in 2001 was assigned to African lineage 1 in the three trees, but presented great changes of phylogenetic positions between the E and NS5 trees. It is mostly close to the strains from Senegal in 1997 in the E tree (Figure 2A) while it clusters with the strains from Coted'Ivoire in 1999 in the NS5 tree (Figure 2B). Although it could not be included in the full-length genomic tree due to insufficient sequence data, strain ArA1465 from Cote d'Ivoire in 1980 was assigned to African lineage 2 in the E tree and African lineage 1 in the NS5 tree. These results suggested that recombination events may have contributed to the phylogenetic differences among ZIKV strains.
The recombination events were evaluated by the RDP software package, which identified three recombination events within the NS5 coding region to generate three recombinants: ArD157995, ArA1456, and ArD158084 (Table 2). Recombination hotspots were identified at nucleotide positions 9652, 9754, 10314, and 10315 (Table 2). The recombinants produced by recombination events 1 and 2, ArD157995 and ArA1456, were identical to the two strains found by visual comparison (>Supplementary Table S1), which further suggested that recombination within NS5 might have resulted in the phylogenetic shifts of ZIKV. Furthermore, each recombinant had major and minor parents, and at least one parent of each recombinant was identified from a different country than the respective recombinant (Table 2). Two of the three recombinants were identified from Senegal, while the other derived from Cote d'Ivoire, and four of the six parents were from Senegal, while one was isolated from Cote d'Ivoire. This indicated that recombination is more likely to occur within the genomes of the ZIKV strains from different lineages co-circulating in Senegal and Cote d'Ivoire, and that migration was more likely to happen between these two countries than among others.
Event
No.Recombinants
(location/time/lineage)Major parents
(location/time/lineage)Minor parents
(location/time/lineage)Methods RDPRCS Begin End 1 ArD157995
Senegal/2001/A1ArA975 Cote
d'Ivoire/1999/A1ArD132915
Senegal/1998/A2GENECONV, MaxChi, RDP, SiScan, 3Seq, Chimaera 0.682 9754 10314 2 ArA1456 Cote
d'Ivoire/1980/A1ArD30101
Senegal/1979/A1ArD132915
Senegal/1998/A2SiScan, 3Seq, Chimaera 0.594 9672 10315 3 ArD158084
Senegal/2001/A1ArB15076 Central African
Republic/1980/A1ArD157995
Senegal/2001/A13Seq, MaxChi 0.685 9672 10315 Note: A1, African lineage 1; A2, African lineage 2. Table 2. The recombination events of ZIKV detected by the RDP software package
-
Migration events and pathways among the ZIKV epidemic countries and territories were characterized (Figure 4). Migration events showing statistical significance (P < 0.01) included one from Brazil to Suriname, two from Cote d'Ivoire to Senegal, and one from Cote d'Ivoire to Uganda (Figure 4, red arrows). Possible migration events (0.01 < P < 0.05) were detected from Senegal as the exporting country, including single events to Nigeria, Malaysia, Philippines, Canada, Cambodia, Thailand, and the Pacific island s, and three events to Cote d'Ivoire (Figure 4, green arrows in dash lines). Therefore, first, Senegal and Cote d'Ivoire are the two countries that have most frequently imported and exported ZIKV, which is consistent with the results of recombination detection showing that the involved strains are mainly from these two countries (Table 2). We speculate that the exchange of ZIKV between Senegal and Cote d'Ivoire might have resulted in the co-circulation of viruses from distinct lineages and thus promoted the recombination of viruses to generate distinct genotypes. Second, Senegal was detected as the exporting country responsible for the possible migration of ZIKV to other countries outside Africa (Figure 4). Thus, Senegal may be the geographic origin of the ZIKV outbreaks outside Africa, which indicates the important role of Senegal in the spread of ZIKV to other countries throughout the history of ZIKV epidemics.
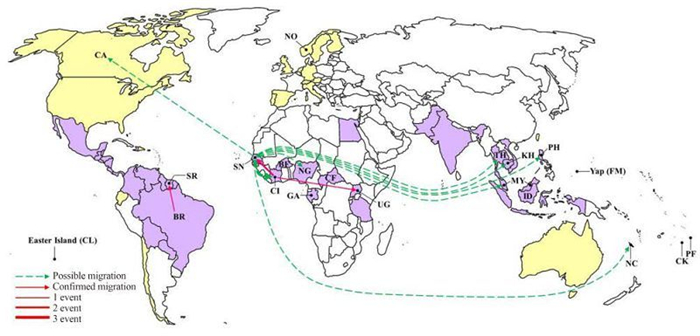
Figure 4. A world map showing the ZIKV migration events and pathways. Countries and areas from which ZIKV strains with complete or partial sequences are available in GenBank are labeled with their abbreviated names as defined in Table 1. The confirmed migration pathways(P < 0.01) from the exporting to the importing countries are indicated by red arrows. The possible migration pathways(0.01 < P < 0.05) are indicated by green arrows in dash lines. The frequency of migration events is illustrated by the thickness of the lines.
Three lineages of ZIKV strains were identified by phylogenetic analysis
The spatial and temporal distributions of the epidemics caused by each ZIKV lineage.
Recombination events in ZIKV indicated potential migration routes
Analysis of the ZIKV migration pathway revealed important roles of Senegal and Cote d'Ivoire in the spread of ZIKV
-
Since the first identification and isolation of ZIKV in 1947 (Dick et al., 1952), records of ZIKV were sparse until the last decade (Kindhauser et al., 2016). Despite the limited numbers of ZIKV sequences available in GenBank, two major lineages of ZIKV, the African and Asian lineages, were characterized in several previous studies (Haddow et al., 2012; Faye et al., 2014). This current study also characterized these two major lineages by constructing a phylogenetic tree for the full-length genome sequences of 23 ZIKV strains (Figure 1). However, in contrast to previous reports on ZIKV genetic relationships based on the limited number of sequences available (Haddow et al., 2012; Faye et al., 2014; Enfissi et al., 2016b), we found that, for the first time, three lineages of ZIKV could be clustered based on the robust E and NS5 trees. These three lineages were designated as the Asian/American lineage, African lineage 1, and African lineage 2, respectively, according to their geographic distributions. Because only partial sequences of the several strains clustered in the additional African lineage 2 were available, this cluster could not be identified by the previous phylogenetic analyses conducted using fewer ZIKV strains.
From our phylogenetic analyses, it is noted that African lineage 2, which has mainly been isolated from Senegal, diverged earlier than the other two lineages, suggesting that the ancestral strains of ZIKV may derive from Senegal. Then, we found that at least two genotypes of ZIKV have co-circulated in Senegal. So, it seems highly probable that recombination between different genotypes has happened in Senegal to generate recombinants with new genotypes. Subsequently, our analysis of recombination events revealed that ZIKV strains from Senegal were more frequently involved in recombination events than other strains. Therefore, strains from Senegal played important roles in ZIKV evolution and divergence. Although it was not so much involved as Senegal, Cote d'Ivoire is also an important country related to ZIKV evolution. Co-circulation of ZIKV strains of different genotypes occurred in Cote d'Ivoire, as African lineage 2, which showed an early divergence from other lineages, included one strain from Cote d'Ivoire (ArA1465) in the E tree (Figure 2A) and other strains from Cote d'Ivoire were clustered in African lineage 1 (Figure 2A and 2B). Moreover, strains from Cote d'Ivoire were also involved in the recombination events. Taken together, our phylogenetic analysis revealed important phylogeographic roles of Senegal and Cote d'Ivoire in ZIKV evolution and divergence.
The migration of ZIKV was elaborated to illustrate the potential pathways for the spread of ZIKV and the geographic origins of recent ZIKV epidemics. Senegal and Cote d'Ivoire were identified to be the countries that had most frequently exported and imported ZIKV strains. Furthermore, ZIKV migrated from Senegal to the Asian countries and Pacific island s where confirmed autochthonous cases were identified, suggesting Senegal as the geographic origin of all known ZIKV epidemics outside Africa. These results are supported by a previous report that illustrated the frequent movement of ZIKV between Senegal and Cote d'Ivoire between 1920 and 1985, and mentioned that the ZIKV lineage from Africa spread to Malaysia around 1945, and to Micronesia around 1960 (Faye et al., 2014). ZIKV export from Senegal directly to Canada was also detected by our analysis using MIGRAPHYLA. However, no autochthonous cases were confirmed in Canada and the patient had recently returned from Thailand (Fonseca et al., 2014). It is possible that the pathway from Senegal to Canada was identified by MIGRAPHYLA because the sequence of the strain PLCalZV that was imported to Canada is mostly similar to that of the strain SV0127/14 from Thailand, which had already imported ZIKV strains from Senegal (Figure 2B). The analyses of ZIKV recombination and migration were conducted according to the phylogenetic analysis in the current study, which are estimated based on the available sequences so far. So the amount of available ZIKV sequences may influence the results of the analyses.
The human cases showed clinical manifestations of ZIKV infections that could be easily mistaken for DEN or CHIK fever. The latter two diseases emerged earlier and were much more commonly diagnosed than ZIKV infections in Africa (Faye et al., 2014). Based on their characterization of the history of ZIKV outbreaks and epidemics globally, Dr. Musso, and his colleagues reported that the spread of ZIKV seems to have followed the same path as DENV and CHIKV and that the potential for ZIKV epidemics might be overwhelming (Musso et al., 2015). The areas at risk of DENV and CHIKV transmission or with identified cases mostly matched with the areas of autochthonous ZIKV outbreaks (Figure 3A). Furthermore, the areas at risk of DENV and CHIKV are all located within the habitat areas of the Ae. Aegypti mosquito (Urdaneta-Marquez and Failloux, 2011), which is one of the principal arthropod vectors of ZIKV (Hayes, 2009), DENV, and CHIKV (World Health Organization, 2014; Weaver and Lecuit, 2015). Although wind-blown mosquitoes can migrate over the sea to distances of several hundred kilometers (Asahina, 1970), it is more likely that the migration of ZIKV over the great distances between continents was achieved as a result of increasing international travel and trade activities by which infected persons and animal hosts were transported to countries that were previously free from ZIKV (Duffy et al., 2009). Further, as the world's human population mostly gathers and lives in areas inhabited by Ae. Aegypti and other mosquitoes, the potential for the worldwide spread of ZIKV is likely to be still increasing. Therefore, it is important to investigate the relationship between ZIKV migration and the distributions and life cycles of mosquito vectors as well as international transport and trade activities. The information that can be provided by such studies will greatly help to prevent and control ZIKV outbreaks worldwide.
-
This work was supported by the Science and Technology Basic Work Program (2013FY113500) from the Ministry of Science and Technology of China.
-
This article does not contain any studies with human or animal subjects performed by any of the authors. The authors declare no conflicts of interest.
-
SS and FD conceived of the study. JMS performed the ZIKV recombination detection and migration analyses. JW downloaded ZIKV sequences available in GenBank and generated the sequence datasets for phylogenetic analysis. ST collected all the information of ZIKV outbreaks and epidemics as listed in Table 1. SS constructed the phylogenetic trees, analyzed all the results, and wrote the first version of the manuscript. HLW, ZHH and FD checked and finalized the manuscript.
-
Strains Locations Time Lineages Full-length E gene NS5 gene ArD157995 Senegal 2001 A1 A1 A1 ArA1465 Cote d'Ivoire 1980 N/A A2 A1 Notes: A1, African lineage 1; A2, African lineage 2; N/A, not applicable. Table Table S1. Strains of discrepancies among tree topology by visual comparison


 DownLoad:
DownLoad: