













HTML
-
Adenoviruses (AdVs), belonging to the family Adenoviridae, are non-enveloped, double-stranded DNA viruses with a genome of 26–45 kb in size. Their genomes are flanked by an inverted terminal repeat (ITR) on each end. At present, five genera in the family Adenoviridae have been identified, including Mastadenovirus, Aviadenovirus, Atadenovirus, Ichtadenovirus, and Siadenovirus (Harrach et al. 2011). Mastadenoviruses infect mammals and aviadenoviruses infect birds (Benkö and Harrach 2003). Atadenoviruses were named because of their unusually high AT content, which have been found in mammals, birds and reptiles (Hess et al. 1997). Siadenoviruses infect amphibians and birds (Davison et al. 2000; Kovács et al. 2010). The Ichtadenovirus genus only contains one species which was isolated from the white sturegeon (Kovács et al. 2003).
AdVs are important, widespread, and occasionally fatal pathogens in humans, as well as wild and domestic animals. So far 103 types of human adenoviruses (HAdVs) have been recognized by The HAdV Working Group (The HAdV Working Group 2019). HAdVs are known to cause cryptic enteric infection, conjunctivitis, acute respiratory disease, hepatitis, and pneumonia (Hiroaki et al. 2008; Matsushima et al. 2013; Ghebremedhin 2014). AdVs are involved both in asymptomatic infections and clinical diseases, whereby virulence, tropism and pathogenesis are typically associated with specific types, and that disease manifestation is additionally host immune status- and agerelated. AdVs have been described in every vertebrate class and in general exhibit a strong species specificity. Interestingly, several reports indicated cross-species transmission of viruses in Mastadenovirus. For example, canine adenoviruses may have originated by interspecies transfer of a bat adenovirus (Kohl et al. 2012). Moreover, it has even been reported that a titi monkey adenovirus (TMAdV) transmitted to human and caused respiratory illness (Chen et al. 2011). Genetic information about adenoviruses is critical to clarify their evolution, transmission and pathogenesis.
Porcine adenoviruses (PAdVs) were first isolated from a rectal swab of a piglet with obvious diarrhea in 1964 (Haig et al. 1964). PAdVs belong to the genus Mastadenovirus and contain 3 species including Porcine mastadenovirus A (PAdV-A), Porcine mastadenovirus B (PAdV-B), and Porcine mastadenovirus C (PAdV-C) (Harrach B 2011). PAdVs were classified into 5 serotypes (PAdV-1 to 5) based on the cross-neutralization test, among which PAdV types 1–3 are closely related, whereas types 4 and 5 are more divergent at the genomic level (Derbyshire et al. 1975; Hayes et al. 1990). PAdV 1–3 belong to PAdV-A. PAdV serotype 4 belongs to PAdV-B. PAdV-5 belongs to PAdV-C. Experimental infections of piglets with PAdV-A are subclinical or associated with intestinal diseases (Sanford and Hoover 1983). PAdV-B causes lesions in several organs including lungs, kidney and brain (Kasza 1966; Shadduck et al. 1967). PAdV-C mainly causes respiratory symptoms such as sneezing with nasal discharges and coughing (Hirahara et al. 1990). Among all the PAdVs, PAdV-B, which was firstly isolated by Kasza et al., is considered as the most pathogenic PAdV and is widely spreading in the United States, Canada, Germany, Hungary, Bulgaria, the United Kingdom, China and the Netherlands (Fig. 1) (Kasza 1966; Elazhary et al. 1985). The full genomic sequence of PAdV-B is critical for the taxonomic and pathogenic studies of these viruses.

Figure 1. Epidemics of PAdV-B in the world. The location where PAdV-B-HNU1 was detected is shown in symbol.
Until now, the complete nucleotide sequences were available for the genomes of PAdV-A, PAdV-C (Reddy et al. 1998; Nagy et al. 2001). For PAdV-B, partial genomic sequences of PAdV-B strain NADC-1 (PVⅢ, E3 13.4 k, E1B and fiber) and strain Kasza (5'ITR) have also been clarified (Kasza 1966; Kleiboeker 1994). Recently, two novel PAdV strains, SVN1 and WI, have been reported, but just partial pol and hexon were identified, thus the classification of these two strains is uncertain (Sibley et al. 2011; Jerman et al. 2014).
The genomic sequence and characteristics, evolutionary status, and tropism of PAdV-B have been the pending problems for a long time. In the present study, we have sequenced the genome of a novel PAdV-B strain, PAdV-BHNU1, and annotated 31 putative genes. This is the first characterization of the complete genomic sequence for PAdV-B. According to the full genome of PAdV-B-HNU1, PAdV-SVN1 and PAdV-WI, two PAdVs with ambiguous taxonomy previously, can be classified into PAdV-B. In addition, the lymphoid tropism of PAdV-B-HNU1 was also preliminarily revealed by the epidemiological data, which may contribute to further pathogenic studies of PAdV-B.
-
To investigate porcine viral pathogens, the porcine samples were collected from pigs showing diarrhea in the Hubei Province, China, during 2017–2018. Totally, 180 lymph nodes, 59 oral swabs and 56 rectal swabs were collected. Virus transport medium (VTM) was made by adding 1% fetal bovine serum (Hyclone) and 1% penicillin–streptomycin (10, 000 U/mL, Gibco) to HBSS medium (Hyclone). The oral and rectal samples were sampled using aseptic swabs and stored in the cryopreservation tubes containing 1 mL VTM. Tissue samples were kept in the cryopreservation tube direct. All the samples were immediately put in dry ice, then transported to the laboratory and stored at –80 ℃.
-
To perform the viral metagenomic analysis, a library was constructed using pooled lymph samples. Briefly, 100 mg tissue of each sample was homogenized using 1 mL phosphate buffer saline, centrifuged, pooled, and filtered. Viral particles in filtrate enriched by ultracentrifugation, and treated with DNase and RNase to digest unprotected nucleic acid. Then viral DNA/RNA was extracted using QIAamp Viral DNA/RNA Mini Kit (Qiagen, Germany) and subjected to random PCR (rPCR) as previously described (Li et al. 2010). The purified rPCR products were used to construct the sequencing library and sequenced on HiSeq-PE150 instrument (Illumina platform). The raw reads were debarcoded, trimmed, and de novo assembled using Geneious software package (Version10.2.2) (Kearse et al. 2012). The assembled contigs were aligned to NCBI nr database using BLASTx with an E-value less than 10-5.
-
For further screening the presence of PAdV-B-HNU1, a set of primers to amplify the partial penton gene (F-5'-GTT AGG CTG TAT GAC TCT GTT GAG-3' and R-5'-CCT AAA CTC CCC ACC CCA GTT AGA TCT C-3'), were designed according the AdV penton gene detected by metagenomic sequencing. Viral DNA was extracted from individual sample with TIANamp Virus DNA Kit (TIANGEN, China) according to the manufacture's instruction. The 15-μL reaction mix contained 2 μL of extracted DNA, 5 pmol each primer (pentonF and R), 7.5 μL 2 × Taq PCR Mastermix (TIANGEN, China). After an initial incubation step at 94 ℃ for 5 min, 40 cycles of amplification were carried out, consisting of denaturation at 94 ℃ for 30 s, annealing at 58 ℃ for 30 s, and extension at 72 ℃ for 40 s, and a final extension step at 72 ℃ for 5 min. PCR products were run in a 1.5% agarose gel. The expected products were cut and extracted using a TIANgel Midi Purification Kit (TIANGEN, China), then sequenced with the two primers.
-
One of the lymph samples positive for PAdV-B-HNU1 was used for genome sequencing. The extracted viral DNA was used as a template for PCR amplification with a combination of primer sets (available upon request) designed for sequencing the genome of PAdV-B-HNU1, according to the metagenomic contig sequences. The expected products were cut and extracted using a TIANgel Midi Purification Kit (TIANGEN, China) and sequenced directly. To confirm the terminal sequences of the viral genome, the end of the extracted viral DNA were tailed using SMARTer RACE 5'/3'Kit (Takara, Japan) and amplified according to the manufacturer's instruction (Elsing and Burgert 1998). PCR products were run by electrophoresis and were sequenced with the genome-specific primers. All the sequences were assembled to a full-length genome.
-
By ORF Finder (National Center for Biotechnology Information), ORFs that code for peptides > 50 aa were identified as potential genes. Gene sequences and encoded putative protein aa sequences were compared to that in the NCBI GenBank using the Basic Local Alignment Search Tool (BLASTn and BLASTp) and the homologous ORFs in other adenoviruses were used to confirm potential genes. Protein identity comparisons were performed by using ClustalW and corrected manually (Thompson et al. 2002). Models of evolution were evaluated using a corrected Aikakeinformation criteria (AICc) in ProtTest3.4.2 to determine the best amino acid substitution model (Darriba et al. 2011). Maximum Likelihood analysis was run in PhyML 3.0 with the best model of evolution according to AICc, using 1000 bootstrap replicates to test the strength of the tree topology (Guindon et al. 2010). Meanwhile, the Neighbor Joining tree was run in MEGA7.0 (Kumar et al. 2016). Trees were edited using FigTree v1.4.3.
-
The following published AdV genome sequences were retrieved from NCBI and included in the analysis in this study: Bat AdV-TJM (NC_016895), Bat AdV-WIV10 (NC_029899), Bat AdV-WIV13 (NC_030874), Bovine AdV-3 (NC_001876), Bovine AdV-2 (NC_002513), California sea lion AdV-1 (NC_024150), Canine AdV-1 (NC_001734), Cervid AdV-1 (NC_030792), Chimpanzee AdV-Y25 (NC_017825), Cynomolgus AdV-1 (NC_034382), Duck AdV-2 (NC_024486), Equine AdV-1 (NC_030792), Fowl AdV-5 (NC_021221), Frog AdV-1 (NC_002501), Goose AdV-4 (NC_017979), Human AdV-1 (AC_000017), Human AdV-54 (NC_012959), Human AdV-7 (NC_004001), Lizard AdV-2 (NC_024684), Murine AdV-3 (NC_012584), Odocoileus AdV-1 (NC_035619), Ovine AdV-7 (NC_004037), Pigeon AdV-2 (NC_031503), Porcine AdV-3 (NC_005869), Porcine AdV-5 (NC_002702), Raptor AdV-1 (NC_015455), Simian AdV-3 (NC_020487), Skunk AdV-PB1 (NC_027708), Squirrel AdV-1 (NC_035207), Titi monkey AdV-ECC-2011 (NC_020487), Tree shrew AdV-1 (NC_004453), Turkey AdV-3 (NC_001958), Turkey AdV4 (NC_022612), Turkey AdV-1 (NC_014564), Porcine AdV-WI (JF699045), Porcine AdV-SVN (KJ933482 and KJ499459).The genome sequences of PAdV-B-HNU1 sequenced in this study were submitted to GenBank (accession no. MK774519).
Sample Collection
Viral Metagenomic Assay
PCR Screening
Full-Length Genome Sequencing
Gene Annotation and Phylogenetic Analysis
Nucleotide Sequence Accession Numbers
-
To investigate the viral infection in the mass lymph node samples collected from pigs, the viral library was constructed with pooled lymph samples of porcine and subsequently sequenced by Illumina platform. The sequencing generated 8, 613, 350 reads with 150 nt length, with an approximate total coverage of 32 ×. After debarcoding and trimming, a total of 6, 673, 308 clean reads were obtained. By de novo assembly, a long contig was obtained from 6, 831 cleaned paired reads. The contig contains genes and encoded proteins that are significantly related to homologues of AdVs in the GenBank. This virus was designated tentatively as PAdV-B-HNU1.
-
To reveal the prevalence of PAdV-B-HNU1 in clinical porcine samples, a set of primers targeting a 349-bp region within the penton base gene, were used to detect PAdV-BHNU1 in 295 porcine samples. The result showed that 108 out of 295 samples (36.6%) were positive for PAdV-BHNU1, including 93 of 180 (51.7%) lymph nodes, 6 of 59 (10.2%) oral swabs and 9 of 56 (16.1%) rectal swabs (Table 1).
Sample types No. of test samples/no. of positive samples (% positive) Lymph 180/93 (51.7) Oral swab 59/6 (10.2) Rectal swab 56/9 (16.1) Total 295/108 (36.6) Table 1. Prevalence of PAdVB-HNU1 in porcine samples collected in China.
-
The full-length genome of PAdV-B-HNU1 was reconstructed by PCR amplification and replicon sequencing using primers designed according to the viral metagenomic contig sequence. The terminal sequences of the viral genome were determined by 5' and 3' RACE amplification and sequencing. After assembly, a 31, 743 bp genome of PAdVB-HNU1 (GenBank no. MK774519) with 55% GC content was obtained.
-
Totally, 116 ORFs (open reading frames) were predicted in both strands of the PAdV-B-HNU1 genome using a searching strategy defining ATG as the start codon and a cutoff size of 50 amino acids (aa). After annotation of the ORFs, the genomic organization of PAdV-B-HNU1 was similar to that of most known members in the genus Mastadenovirus (Fig. 2). The GC contents vary from 46 to 65% in different genes, among which genes in the center of the genome, stretching from IVa2 to pVⅢ, showed higher GC contents than the genes at the terminal regions. Among all the putative proteins encoded by PAdV-B-HNU1, the aa sequences of 31 products were homologous to those of other AdVs, of which 21 were located at the positive strand and 10 were located at the complementary strand (Table 2, Fig. 2). Next, genes of PAdV-B-HNU1 were classified into two sets, homologous genes in all genera as 'genus-common genes' and other genes as 'genus-specific genes' (Davison et al. 2003). Similar to other members of the genus Mastadenovirus, the middle part of the PAdV-BHNU1 genome was predicted to contain 18 genus-common genes. These include genes coding for protein related to DNA replication (Pol, pTP, and DBP), DNA encapsidation (52 K and Iva2), virion formation and capsid structure (pⅢa, penton base, pVⅡ, pX, pVI, hexon, protease, 100 K, 33 K, pVⅢ, and fiber); 22 K, which originates from a lack of splicing in 33 K; and U exon which has been lost in PAdV-C. And PAdV-B-HNU1 had the longest inverted terminal repeat compared to other porcine adenoviruses (Fig. 3).

Figure 2. Genome organization of PAdV-B-HNU1 and other adenoviruses. The viral genome is represented by the thick line in the center marked with 10-kb intervals. Rectangles represent genome regions of PAdV-B published online and the dashed line indicates unavailable region of PAdV-B. Arrows underneath the genome line predicted ORFs of adenoviruses and the thin lines indicate the introns.
Gene product Location(s) (nt)a Size (aa) Amino acid identity (%) with: GC content (%) PAdVNADC1 C.sea lion AdV1 PAdVA PAdVC E1 ORF 1 Termini 485–1039 186 94 29 22 23 55 E1B 19 kDa 1762–2220 152 NI 26 14 NI 54 E1B 55 K 2127–3368 413 95 38 24 28 64 IX 3466–3801 111 100 29 24 26 56 184R 3981–4280 99 NA NI 50 NI 48 ORF6 26, 760–27, 041 93 96 NI NI NI 46 ORF7 26, 242–26, 760 172 97 NI NI NI 56 ORF4 27, 022–27, 561 119 99 NI NI NI 49 U exon 27, 560–27393c 55 100 53 38 NI 48 Fiber 27, 575–29, 764 729 94 28 14 14 55 E434kDa 30, 753–30004c 249 NA 36 28 26 51 dUTPase 31, 592–31164c 145 NA 50 NI 40 62 E415kDa 31, 151–30750c 133 NA 30 NI NI 53 IVa2 Center 3891–5218, 5298–5310c 373 NA 69 58 65 50 pol 4982–8410, 12, 937–12966c 1037 NA 69 65 55 55 11.5 kDa 6087–6407 106 NA NI NI NI 54 p52k 10, 128–11, 339 403 NA 61 47 48 56 Penton base 13, 035–14, 483 482 NA 76 60 63 53 pV 15, 234–16, 175 313 NA 28 14 23 61 pX 16, 197–16, 391 64 NA 70 48 59 57 pVI 16, 428–17, 159 244 NA 48 34 44 61 pTP 8263–10, 095, 13, 097–13162c 595 NA 72 55 58 59 pⅢa 11, 266–12, 927 554 NA 62 43 48 58 p33K 24, 249–24, 423, 24, 690–25, 000 161 NA 52 29 NI 64 p22K 24, 625–24, 912 95 NA 48 29 NI 55 pVⅡ 14, 486–15, 178 230 NA 48 27 24 65 Hexon 17, 201–20, 161 986 NA 77 63 66 57 protease 20, 165–20, 791 208 NA 64 63 57 54 DBP 22, 379–20853c 508 NA 55 41 39 65 p100K 22, 394–24, 403 669 NA 64 50 54 58 pVⅢ 25, 031–25, 696 221 94 57 50 52 62 NA not applicable due to the lack of detectable similarity, NI homologous gene not identified.
aThe letter "c" in this column indicates that those genes are encoded by the complementary strand.Table 2. Putative proteins encoded by PAdV-B-HNU1.

Figure 3. Comparison of nucleotide sequences of 5'UTR between PAdV-B-HNU1, PAdV-C, PAdV-A and C. sea lion AdV-1. Pairwise alignments were calculated using DNAMAN. Dots indicate gaps.
A summary of the sequence identity among proteins of PAdV-B-HNU1 and other porcine adenoviruses including California sea lion AdV-1 is presented in Table 2. The amino acid sequences (94–100%) of PAdV-B-HNU1 showed the highest similarity to PAdV-B strain NADC-1 compared to other porcine adenoviruses including PAdV-A and PAdV-C, suggesting that PAdV-B-HNU1 belongs to PAdV-B.
-
Several PAdV strains, such as PAdV-SVN1 and PAdV-WI, have not been classified into any species, because only part of their genome has been sequenced for which the speciesreference sequence is lacking. The PAdV-B-HNU1 provides the first representative complete genomic sequence of PAdV-B, which can be used to classify related PAdV strains. The classification of the PAdV-SVN1 and PAdVWI is made by comparing the aa sequence identities. According to multiple alignment of partial DNA polymerase (Fig. S1A) and hexon (Fig. S1B) aa sequence, partial DNA polymerase of PAdV-SVN1 shows 99%, 76%, and 74% identity to PAdV-B-HNU1, PAdV-A, and PAdVC, respectively, while partial hexon of PAdV-WI shows 93%, 93%, 56%, and 56% identity to PAdV-SVN1, PAdVB-HNU1, PAdV-A, and PAdV-C, respectively. This suggests that PAdV-SVN1 and PAdV-WI, the two PAdVs whose taxonomic position was uncertain, belong to PAdVB.
-
Phylogenetic analysis was conducted to reveal the genetic and evolutionary status of PAdV-B-HNU1. Phylogenetic trees were constructed based on the amino acid sequences of DNA polymerase, hexon, penton base and pre-terminal protein. In these trees, the PAdV-B-HNU1 strain was clustered with C.sea lion AdV-1 with high bootstrap value, indicating that PAdV-B-HNU1 is a member of the genus Mastadenovirus (Fig. 4, Fig. S2). And the genome of PAdV-B-HNU1 shared highest nucleotide sequence identity (54.1%) with C. sea lion AdV-1 (Table S1).
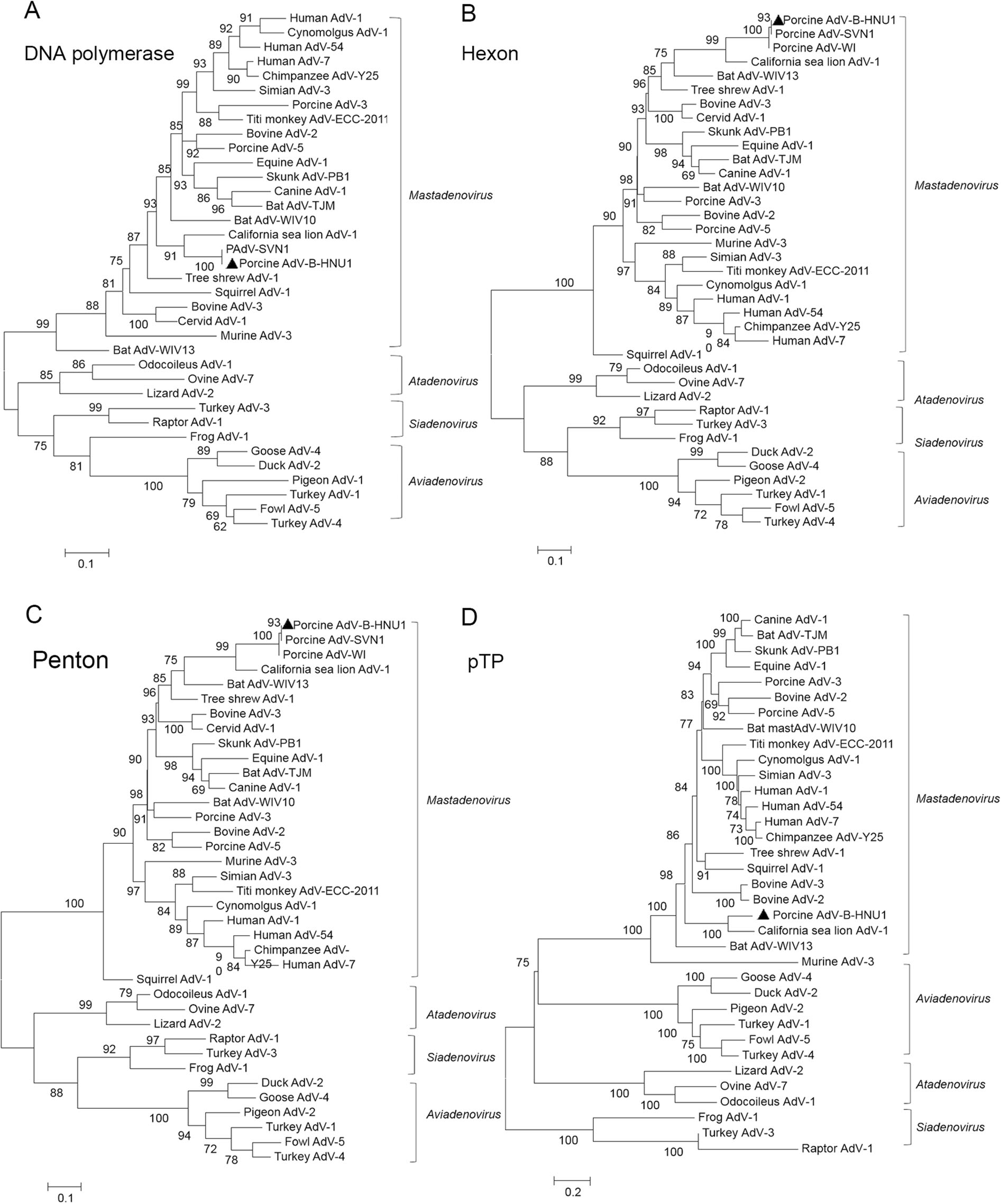
Figure 4. The evolutionary position of PAdV-B-HNU1 in the phylogenetic tree of adenoviruses. The phylogenetic trees were constructed based on the amino acid sequences of DNA polymerase (A), hexon (B), penton base (C) and pTP (D) of porcine adenoviruses by using the maximum likehood (ML) method with rtREV + F + I, LG + F + R5, LG + I + G + F, and LG + G as the model of protein evolution, respectively. PAdV-B-HNU1 detected in this study was indicated with solid triangle.
A dUTPase was identified at the 3' end of PAdV-BHNU1 genome, which was more similar to dUTPases of bacteria and fungi than that of mastadenoviruses except C. sea lion AdV-1. Phylogenetic analysis further confirmed that PAdV-B-HNU1 dUTPase does not cluster with other mastadenoviral dUTPases and was more related to dUTPases of baculovirus, poxvirus and eukaryotes (Fig. 5). This suggested that PAdV-B-HNU1 is more closed to C. sea lion AdV-1 than other PAdVs in evolutionary positions.
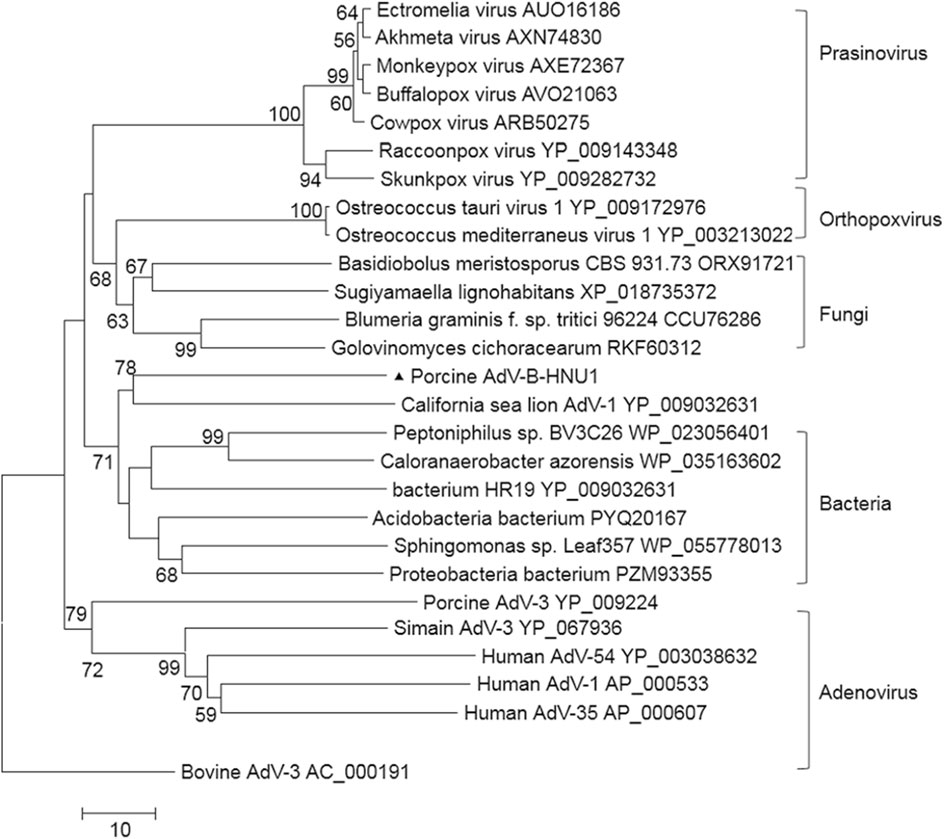
Figure 5. Phylogenetic tree constructed on the amino acid sequences of dUTPase. Phylogenetic analysis of PAdV-B-HNU1 is based on the partial conserved amino acid sequences of dUTPase from selected adenoviruses, bacteria, fungi, prasinovirus and poxviruses by using the Maximum Likelihood method based on the JTT matrix-based model. PAdV-B-HNU1 detected in this study was indicated with solid triangle.
Detection of a Novel HNU1 Strain of PAdV-B by Viral Metagenomics
Prevalence of PAdV-B-HNU1 in Porcine
Characterization of the Complete Genome of PAdV-B-HNU1
Genome Analysis of PAdV-B-HNU1
Classification of PAdV-SVN1 and PAdV-WI Based on PAdV-B-HNU1 Genomic Sequence
Phylogenetics of PAdV-B-HNU1
-
Three PAdVs species, PAdV-A, PAdV-B and PAdV-C, have been established based on genomic features and phylogeny. Currently, the genomes of PAdV-A and PAdVC have been completely sequenced and well annotated, but the genome of PAdV-B has not been identified yet. Here we presented the first complete genomic sequence of PAdV-B-HNU1, a novel strain of PAdV-B.
Sequence analysis of PAdV-B-HNU1 genome indicated that the overall genomic structure of PAdV-B-HNU1 is similar to that of most known AdVs. Phylogenetic analysis based on the DNA polymerase sequence and the penton base protein sequence classified PAdV-B-HNU1 into the genus Mastadenovirus, and showed that PAdV-B-HNU1 is most closely related to C. sea lion AdV-1. However, the average GC content of PAdV-B-HNU1 was 55%, higher than that of C. sea lion AdV-1 (36%). In different genes of PAdV-B-HNU1, the GC content ranged from 46% to 65%, with high rates in the center of the genome and low rates at terminal regions. High abundance of 5'-C-phosphate-G-3' (CpG) in the genomes was responsible for GC contents (Tan et al. 2017). The center of adenoviruses genome contained structural proteins and enzymes of which high expression levels were required for DNA replication, DNA encapsidation and the formation of the virion (Hoeben and Uil 2013). High GC content in certain ORFs may increase CpG islands, affect viral DNA methylation and subsequently modulate gene expression. However, this speculation lacked any evidence currently and we would focus on it in our future studies. The ITRs played an important role in the initiation of replication by providing binding sites for viral and cellular regulatory proteins in adenoviral infection (Dán et al. 2001). The first 17 nucleotides of this region were identical in PAdV-5 and PAdV-3, but they are less conserved in PAdV-B-HNU1.
Fiber protein exists in all adenoviruses for tethering of the viral capsid to the cell surface via its interaction with the cellular receptor such as CAR (Roelvink et al. 1998), or CD46/80/86 (Short et al. 2006), or sialic acid (Arnberg et al. 2000). The carboxy-terminal end of the predicted fiber of PAdV-B-HNU1 showed similarity to galectin-4 and galectin-9 of animals, which was similar to PAdV NADC-1 having a fiber containing a C-terminal galectin domain (Guardado-Calvo et al. 2010). Galectin-4 plays an important role in lipid raft stabilization, protein apical trafficking, cell adhesion, wound healing, intestinal inflammation, tumor progression (Cao and Guo 2016). Galectin-9 can interact with CD40 on T-cells induced their proliferation inhibition and cell death (Vaitaitis and Wagner 2012). Whether the PAdV-B-HNU1 fiber gene plays a similar role remains to be determined.
dUTPases were ubiquitously expressed enzymes in eukaryotic and prokaryotic cells as well as viruses. dUTPase catalyzes the conversion of dUTP to dUWP and PPi, reducing dUTP/dTTP ratio to prevent misincorporation of uracil into DNA (Koonin 1996). Viral dUTPases were captured by some viruses via horizontal gene transfer (Baldo and Mcclure 1999). Viral dUTPases showed species specificity and have been discovered in herpesviruses (Glaser et al. 2006), poxviruses (Cottone et al. 2002) and retroviruses (Payne and Elder 2001). The dUTPase activity has been confirmed in the case of fowl adenovirus 1, and fowl adenovirus 9 dUTPase upregulated the expression of type I Interferons (Weiss et al. 1997; Deng et al. 2016). PAdV-B-HNU1 dUTPase was more divergent compared to those of other PAdVs. Similar to C. sea lion AdV-1, the predicted PAdV-B-HNU1 dUTPase was more closely related to dUTPase of bacteria and fungi than those of mastadenoviruses. This indicated a potential independent horizontal gene transfer event that bacterial dUTPase may be an endogenous viral element derived from adenovirus. Whether the PAdV-B-HNU1 dUTPase shares similar biological activity like other adenoviruses remains to be determined.
The genomic information for fiber and dUTPase is not available for all porcine adenoviruses. We proposed the HNU1 strain as the reference strain of PAdV-B. Porcine adenovirus strain PAdV-WI which was detected in pen wash water of newborn to finisher pigs and was only available for a fragment of hexon gene, has been proposed as a prototype of a new species in the Mastadenovirus genus (Sibley et al. 2011). In addition, PAdV-SVN1 was detected in urinary bladder urothelial cell culture and was available for partial polymerase and hexon (Jerman et al. 2014). Limited phylogenetic analysis showed that PAdVSVN1 clustered together with PAdV-WI, that was clearly separated from other Mastadenovirus representative strains (Jerman et al. 2014). The clear classification of PAdVSVN1 as well as PAdV-WI is not possible until the whole genome sequence of PAdV-B-HNU1 is available. According to the amino acid sequence of the DNA polymerase and hexon in this study, the PAdV-SVN1 and PAdV-WI are closely related to HNU1 strain. We proposed PAdV-SVN1 and PAdV-WI can be classified into PAdVB. With the completion of full genomes from all porcine adenovirus species, additional insight into the classification and evolution were obtained.
C. sea lion AdV-1 caused acute hepatitis in sea lions and other fins (Goldstein et al. 2011; Inoshima et al. 2013), which was predicted to jumped to California sea lions from an unknown mammalian endemic host (Cortéshinojosa et al. 2015). Similarities between PAdV-B-HNU1 and C. sea lion AdV-1 were apparent not only in their genome organization, but also in their phylogenetic relationship. Phylogenetic analyses showed that members of PAdV-B were closely related to C. sea lion AdV-1 with the similar dUTPase gene. Further investigation of infectious characters of C. sea lion AdV-1 and PAdV-B-HNU1 was blocked by the failure of virus isolation. More studies, such as infecting swine cells and infecting sea lion cells with C. sea lion AdV-1 and PAdV-B-HNU1, respectively, will contribute to better understanding of the and transmission of these viruses.
In all, our study on PAdV-B-HNU1 has provided the first representative sequence and annotation of PAdV-B genome, contributing to the classification of PAdVs. For instance, PAdV-SVN1 and PAdV-WI, two unclassified PAdV strains, can be classified into PAdV-B species comparing to the genomic sequence of PAdV-B-HNU1. In addition, the PAdV-B-HNU1 was probably a lymphotropic adenovirus, which may explain the specific pathogenic pattern of PAdV-B-HNU1 infection. Considering that PAdV-Bs widely spread in different countries around the world, our findings would contribute to the future epidemic studies of PAdV-B.
-
This work was jointly funded by the National Key Research and Development Program of China (grant number 2017YFD0500104), the Hu-Xiang Youth Talents Scholar Program of Hunan Province (grant number 2017RS3017), the Science Fund for Distinguished Young Scholars of Hunan Province (grant number 2019JJ20004), National Natural Science Foundation of China (grant number 81902070), the Provincial Natural Science Foundation of Hunan Province (grant number 2019JJ50035) and the Fundamental Research Funds for the Central Universities of China (grant number 531107051162).
-
Conceptualization, SL and XG; Methodology, SL, QW, TL, SZ, JL, YQ and XG; Investigation, SL, QW, TL, SZ, JL, LW, YQ and XG; Writing—Original Draft, SL; Writing—Review & Editing, SL, YQ and XG; Funding Acquisition, YQ and XG; Resources, TL, SZ, LW and XG; Supervision, YQ and XG.
-
The authors declare that there are no conflicts of interest.
-
All institutional and national guidelines for the care and use of laboratory animals were followed.


 DownLoad:
DownLoad: