














HTML
-
Porcine reproductive and respiratory syndrome (PRRS), first occurred in the United States in 1987, is an endemic disease causing huge economic losses to pig farms around the world (Garner et al. 2001; Neumann et al. 2005; Rossow 1998). PRRSV, the causative agent of PRRS, belongs to the family Arteriviridae in the order Nidovirales (Cavanagh 1997). It exists as two distinct virus species, that is, PRRSV-1 and PRRSV-2 (Adams et al. 2017; Wang et al. 2020). The PRRSV genome is a single-stranded positive-sense RNA, about 15 kb in length with 10 open reading frames (ORF) encoding eight structural proteins and at least 14 non-structural proteins (nsps) (Chand et al. 2012; Dokland 2010; Meulenberg 2000; Montaner-Tarbes et al. 2019). The clinical outcomes of PRRSV infection are characterized by severe reproductive failure in pregnant sows, pneumonia in piglets, and increased mortality of pigs at all ages (Music and Gagnon 2010).
Transcription factor DP2 (TFDP2), first isolated in 1995, forms heterodimers with the E2F family of transcription factors (Rogers et al. 1996; Wu et al. 1995). The binding of DP2 to E2F potentiates the DNA affinity and transcriptional activity of E2F (Wu et al. 1995; Zhang and Chellappan 1995). The target genes of E2F/DP heterodimers are implicated in a variety of cellular processes including cell cycle progression, DNA repair, cell differentiation, and apoptosis (Hitchens and Robbins 2003; Lam and La Thangue 1994). The eukaryotic cell cycle can be divided into four stages: G1, S, G2 and mitosis (M) (Wenzel and Singh 2018). It has been reported that some viruses can affect host cell cycle progression to provide a cellular environment that is advantageous for viral proliferation (Balistreri et al. 2016; Grey et al. 2010; Laichalk and Thorley-Lawson 2005; Swanton and Jones 2001). The cell cycle progression is tightly regulated by the binding of cyclins and cyclin-dependent kinases (CDKs) (Morgan 1995). It has been shown that PRRSV can arrest the cell cycle of MARC-145 cells (Song et al. 2018). However, how PRRSV manipulates the cell cycle to facilitate its proliferation is still unclear.
In this study, the mRNA expression profiles of transcription factors in PRRSV-infected porcine alveolar macrophage 3D4/21 cells were analyzed. The transcription factor TFDP2 was found to increase in PRRSV-infected macrophages, and in turn, it facilitated the PRRSV proliferation. The relationship between TFDP2 and PRRSV proliferation was further investigated. Our study provides new ideas for the prevention and control of PRRSV.
-
Porcine alveolar macrophage (PAM) cell lines CRL2843-CD163 (3D4/21) were kindly provided by Prof. Jun Han from China Agricultural University and maintained in RPMI-1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% (V/V) fetal bovine serum (FBS, Biological Industries, Kibbutz Beit-Haemek, Israel) and 100 U/mL penicillin, and 100 μg/mL streptomycin. Human embryonic kidney (HEK) 293 T cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% FBS. All cells were maintained at 37 ℃ with 5% CO2. HP-PPRSV JXwn06 was used in this work and the titer was identified to be 104 PFU/mL as previously described (Su et al. 2018).
Anti-PRRSV Nsp2 monoclonal antibody was kindly contributed by Prof. Jun Han from China Agricultural University. A polyclonal antibody against TFDP2 was generated in our laboratory. anti-FLAG antibody and anti-p–C/EBPβ antibody were purchased from Cell Signaling Technology (CST, Boston, MA, USA). Anti-C/EBPβ antibody was purchased from ImmunoWay Biotechnology (Plano, TX, USA). Anti-β-actin antibody and secondary antibodies were purchased from Invitrogen (Carlsbad, CA, USA).
-
3D4/21 cells were seeded in 6-well plates and mock-infected or infected with 0.5 MOI PRRSV for 24 h. Then cells were washed twice by PBS and added 1 mL Trizol reagent (Invitrogen, Carlsbad, CA, USA). The treated cells were sent to the Guangzhou GENE DENOVO Company for RNA sequencing. Data and a heatmap of differentially expressed genes were analyzed by Heml software.
-
The gene encoding TFDP2 (GenBank Accession No. XM_021069556.1) was amplified from 3D4/21 cells cDNA using the primers listed in Supplementary Table S1 and cloned into pGEM®-T Easy Vector (Transgen, Beijing, China). The CDS region of TFDP2 was amplified by PCR using specific primer pairs (Supplementary Table S1) and subcloned into the pFlag-CMV2 vector using a one-step clone kit (Vazyme, Nanjing, China).
Genomic DNA was extracted from PAMs using a DNA extraction kit (TaKaRa, Otsu, Shiga, Japan). The 1701-bp porcine TFDP2 promoter sequence (NC_010455.5) relative to the transcription initiation site (+1) was amplified by specific primers and ligated into luciferase reporter vector pGL3-Basic (named − 1301/400-Luc) using a one-step clone kit (Vazyme, Nanjing, China). The truncated mutants of TFDP2 promoter were then constructed by PCR using the − 1301/400-Luc plasmid as a template (− 792/400-Luc, − 67/400-Luc, − 17/400-Luc). Site-directed mutagenesis of the C/EBP-β, ATF-1, AP-1, SP1 binding sites was performed by PCR using the − 17/400-Luc vector as a template. The 1291-bp porcine cyclin A promoter (NC_010450.4) luciferase reporter plasmid was constructed as above described. The pRL-TK Renilla luciferase reporter plasmid was used as an internal control. All primers are listed in Supplementary Table S1.
-
Total RNA was extracted from 3D4/21 cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and the first-strand cDNA was synthesized using a First-Strand Synthesis System (Transgen, Beijing, China) according to the manufacturer's instructions. The relative gene expression was analyzed by real-time RT-qPCR carried out on qTOWER3G Real-time PCR system (Analytik Jena AG, Jena, Germany). Relative gene expression levels were calculated using the comparative cycle threshold (CT) method according to the manufacturer's protocol (Applied Biosystem, Foster City, CA, USA). All data presented are relatively quantitative, normalized to the β-actin and analyzed using GraphPad Prism 8.0 software. All primers used for real-time RT-qPCR are listed in Supplementary Table S2.
-
Cells were lysed in RIPA buffer (Solarbio, Beijing, China) with 100 U of proteinase inhibitors phenylmethanesulfonyl fluoride [PMSF] (Solarbio, Beijing, China). Cellular proteins were separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). Membranes were then blocked with 5% skim milk in TBST (0.05% Tween-20) for 1 h and incubation overnight at 4 ℃ with the antibodies anti-PRRSV Nsp2 (1:1000), anti-TFDP2 (1:500), anti-p–C/EBPβ (1:1000), anti-C/EBPβ (1:1000), anti-β-actin (1:5000) or anti-FLAG (1:5000). Then the membranes were washed with PBST three times and incubated with HRP-conjugated secondary antibodies (1:5000) for 1 h. Signals were visualized using Pierce ECL Western Blotting Substrate (Thermo Scientific, Wilmington, DE, USA).
-
3D4/21 or HEK293T cells were seeded in 24-well plates and transfected with the constructed plasmids [pRL-TK (20 ng), pGL3-Basic or cyclin A/TFDP2 promoter mutant plasmids (200 ng)] using Lipofectamine 3000 reagent (Invitrogen). At 24 h post-transfection, the cells were infected with or without PRRSV at a multiplicity of infection (MOI) of 0.5 for 24 h. The lysed samples were prepared and analyzed for firefly and Renilla luciferase activities using a dual-luciferase reporter assay system (Yeasen, Shanghai, China) following the manufacturer's instructions.
-
Small interfering RNAs (siRNAs) targeting TFDP2 (siTFDP2) and C/EBPβ (siC/EBPβ) genes or negative control (NC) were designed and synthesized by GenePharma (Shanghai, China) (Table S3). Briefly, 3D4/21 cells were seeded in 12-well plates and transfected with siRNAs at a final concentration of 50 nmol/L using Lipofectamine 3000 (Invitrogen). Then cells were infected with 0.5 MOI PRRSV for 24 h. The gene expression levels were assessed by real-time RT-qPCR and Western blotting.
-
3D4/21 cells were cultured in RPMI-1640 medium supplemented with 10% FBS and then cultured for 24 h in serum-free supplemented medium (cells arrested in the G0/G1 phase). 3D4/21 cells were seeded in 6-well plates until the cell density was about 80% following treatment with thymidine (2 mmol/L, Sigma, St. Louis, MO, USA), nocodazole (80 ng/mL, Selleckchem, Houston, TX, USA) for 24 h (cells arrested in the S, G2/M phase, respectively).
-
For cell cycle analysis, 3D4/21 cells were seeded in 12 well plates and transfected with 1 μg Flag-TFDP2 or vector, or transfected with siTFDP2 or NC at a final concentration of 50 nmol/L. At 12 h post-transfection, the cells were infected with PRRSV at 0.5 MOI. The cells were harvested and fixed with precooled 70% ethanol at 4 ℃ overnight. Then the cells were incubated with propidium iodide (PI) in the dark for 30 min. The DNA content was analyzed by flow cytometry (BD Biosciences, San Jose, CA, USA).
-
3D4/21 cells in different cell cycles were infected with PRRSV at an MOI of 0.5. At 24 h postinfection, cells were fixed with 4% paraformaldehyde and permeabilized with phosphate-buffered saline (PBS) containing 0.3% Triton X-100 for 10 min. Then cells were blocked with 5% BSA–PBS for 30 min and stained with anti-Nsp2 antibody for 1 h at room temperature. Following a wash performed with PBS three times, cells were incubated with FITC-conjugated goat anti-rabbit IgG secondary antibody for 1 h. Nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI) (Molecular Probes, Waltham, MA, USA). Immunofluorescence was observed with an Olympus confocal microscope. Images were taken at × 100 magnification.
-
The virus titer was determined using the 50% tissue culture infective dose (TCID50) method as previously described (Zhang et al. 2018).
-
All experiments were performed with at least three independent replicates. Data were analyzed by GraphPad Prism software (version 8.0) and differences were analyzed using Student's t-test. A P value < 0.05 was considered statistically significant.
Cells, Virus, and Antibody
RNA-Sequencing
Plasmid Construction
Quantitative Real-Time Reverse-Transcription PCR (real-time RT-qPCR)
Western Blot Analysis
Luciferase Assay
RNA Interference
Cell Cycle Synchronization
Flow Cytometry Analysis
Confocal Immunofluorescence
Determination of Virus Titer
Statistical Analysis
-
To screen for genes whose transcription was significantly changed in PRRSV-infected 3D4/21 cells, RNA high throughput sequencing (RNA-Seq) was performed. The mRNA expression profiles of transcription factors showed an up-regulation of TFDP2 after PRRSV infection (Fig. 1A). To further validate the results of the RNA-Seq data, 3D4/21 cells were infected with 0.5 MOI PRRSV. Then total RNA was extracted and the mRNA level of TFDP2 was detected by real-time RT-qPCR. The results showed that PRRSV infection remarkably induced TFDP2 mRNA expression (Fig. 1B). Consistently, Western blot analysis also showed the increased protein level of TFDP2 after PRRSV infection (Fig. 1C). Together, these data demonstrate that PRRSV infection upregulates TFDP2 production in 3D4/21 cells.
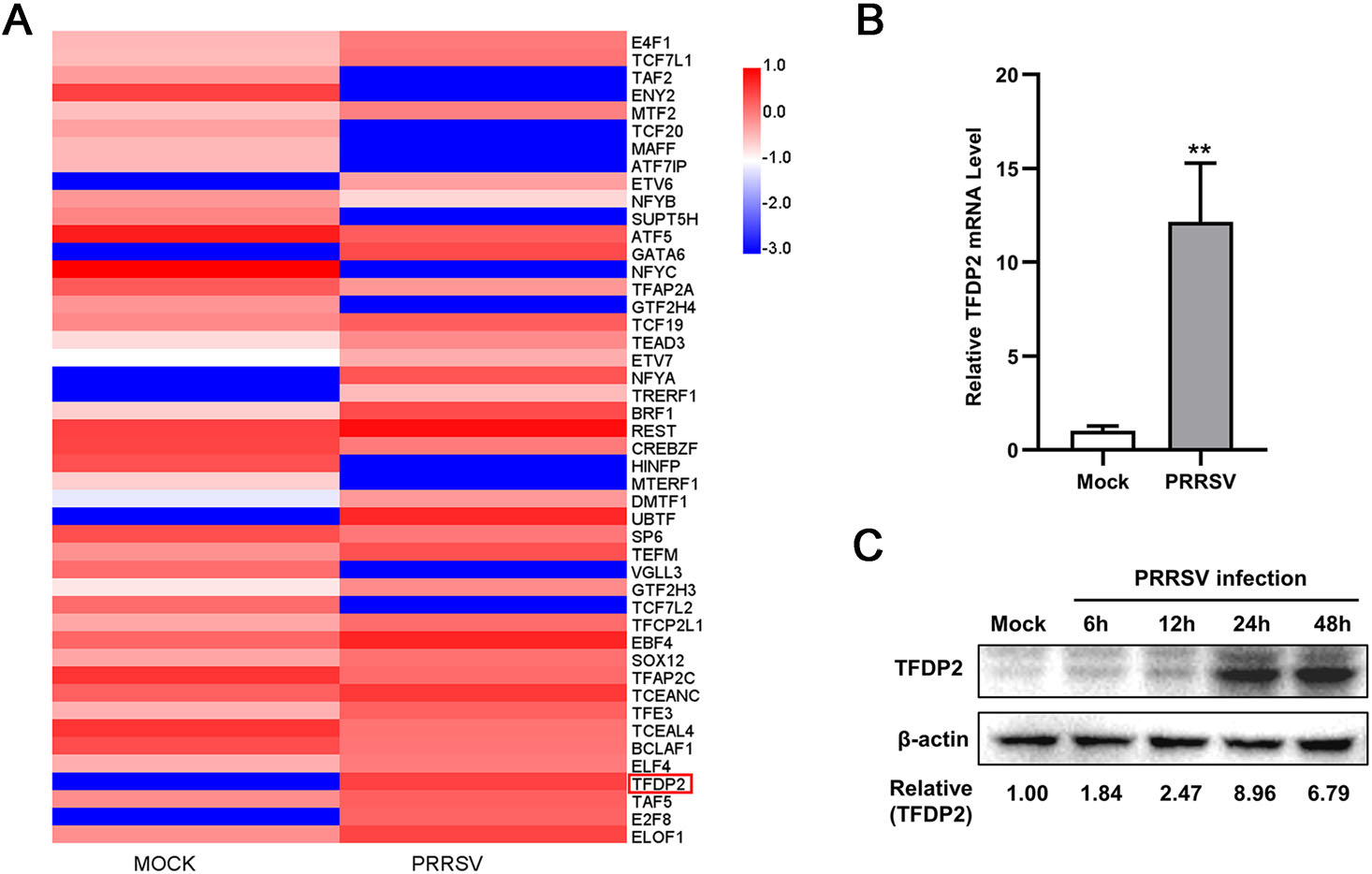
Figure 1. TFDP2 is upregulated in 3D4/21 by PRRSV. A The heatmap of transcription factors-related differentially expressed genes. B Real-time RT-qPCR analysis of TFDP2 in 3D4/21 cells infected without or with 0.5 MOI PRRSV for 24 h. C 3D4/21 were mock-infected or infected with 0.5 MOI PRRSV. At the indicated time points, cells were harvested and subjected to Western blotting (WB) to analyze the expression of TFDP2. Relative expression levels shown below the images were evaluated as fold changes after normalization to β-actin levels. Data are representative of three independent experiments. Significant differences are indicated as follows: **, P < 0.01.
-
To figure out if the expression of TFDP2 affects PRRSV proliferation, we overexpressed TFDP2 in 3D4/21 cells and then inoculated with PRRSV for 24 h. It was found that overexpression of TFDP2 increased the mRNA level of the PRRSV open reading frame 7 (ORF7) (Fig. 2A). The results of Western blot analysis showed the increased Nsp2 protein level in TFDP2-transfected cells (Fig. 2B). Meanwhile, the virus titer was higher in cells overexpressing TFDP2 than that of control (Fig. 2C).
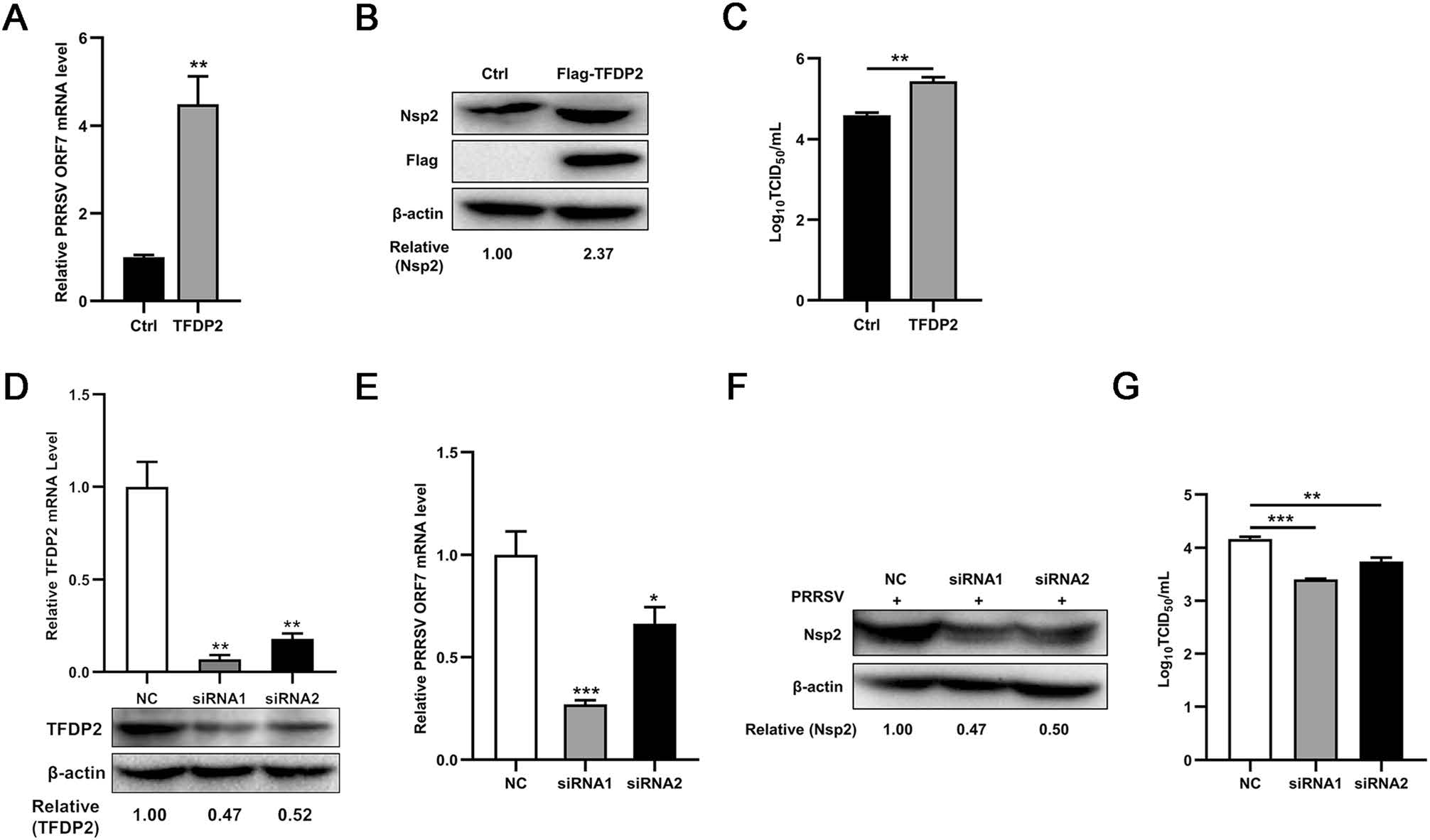
Figure 2. TFDP2 contributes to PRRSV proliferation. A−C Flag-TFDP2 plasmid or empty vector were transfected into 3D4/21 cells, after 12 h post-transfection, cells were infected with 0.5 MOI PRRSV and were collected at 24 h. PRRSV ORF7 level was detected by real-time RT-qPCR (A), and PRRSV Nsp2 was shown by Western bolt (B). PRRSV titers in supernatants were tested by TCID50 assay (C). D−G TFDP2 siRNAs interference effects were detected by real-time RT-qPCR and Western blot (D). PRRSV ORF7 was detected by real-time RT-qPCR (E), and PRRSV Nsp2 levels were tested by Western blot (F). PRRSV titers in supernatants were tested by TCID50 assay (G). Data are representative of three independent experiments. Significant differences are indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further clarify the above results, we designed two small interfering RNA (siRNA) sequences targeted to TFDP2, and the efficiencies of interference were identified by real-time RT-qPCR and Western blot (Fig. 2D). As shown in Fig. 2E−2G, PRRSV proliferation was suppressed when TFDP2 knockdown. Taken together, these data suggest that TFDP2 facilitates PRRSV proliferation.
-
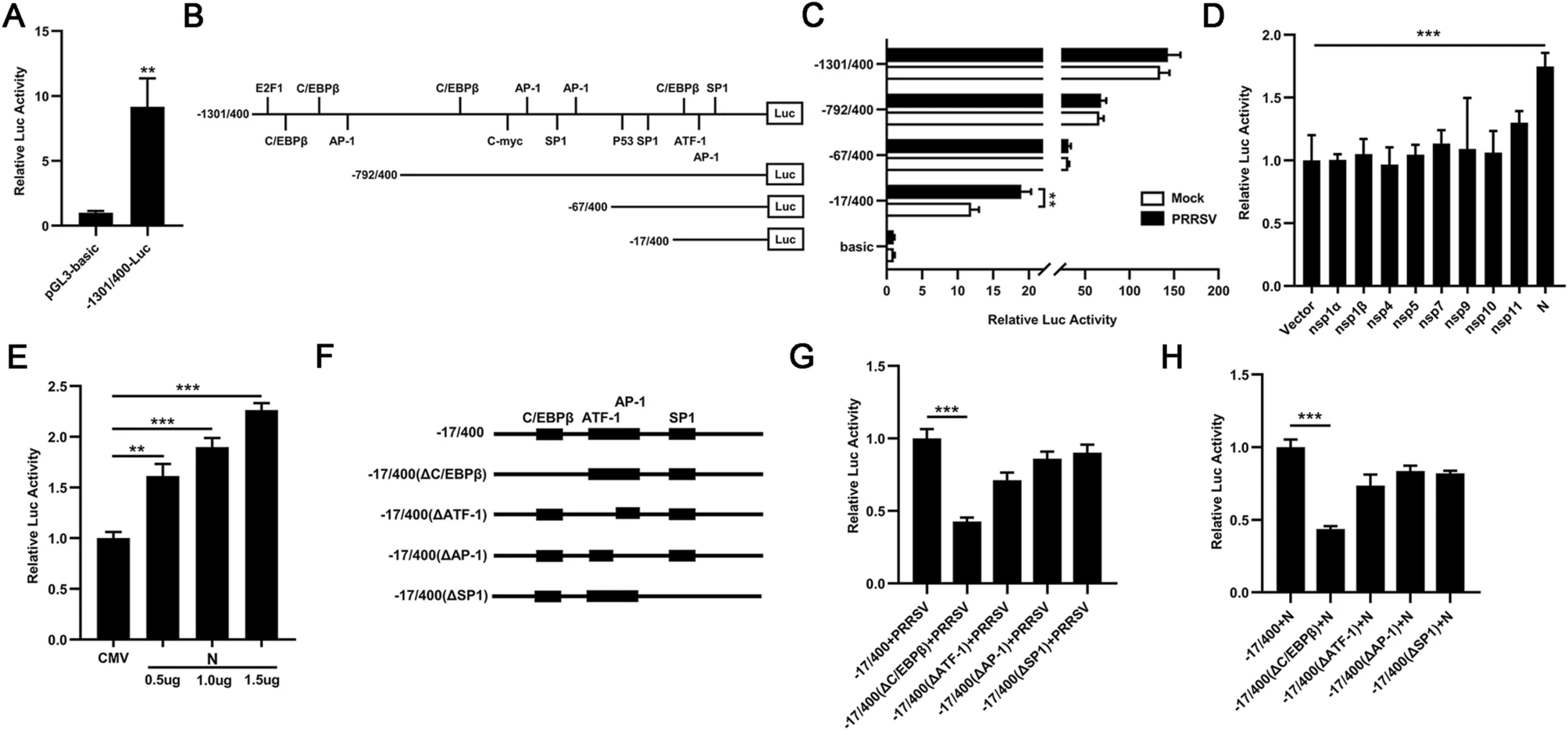
To investigate the mechanism by which PRRSV induced the transcription of TFDP2, a 1701 bp fragment of the 5′ flanking region of the porcine TFDP2 gene was cloned to construct the TFDP2 promoter-reporter plasmid −1301/400-Luc. And the activity of TFDP2 promoter was identified by luciferase assay (Fig. 3A). Using the bioinformatics approach (http://alggen.lsi.upc.es) (Bi et al. 2014), we identified several putative transcriptional regulatory elements on the promoter of the TFDP2 gene, and constructed a series of promoter truncated mutants schematically shown in Fig. 3B. To determine the region of the TFDP2 promoter that is responsive to PRRSV infection, 3D4/21 cells were transfected with these constructs. Twenty-four hours after transfection, the cells were infected or mock-infected with 0.5 MOI PRRSV. The luciferase activities of the −17/400-Luc truncated mutant showed 1.6-fold upregulation upon PRRSV infection, while other mutants did not (Fig. 3C). These results indicate that regulatory elements might exist in the TFDP2 promoter region from −17 to +400 bp and the responsive elements could be the putative C/EBPβ (−11 to −4), ATF-1 (+30 to +40), AP-1 (+37 to +43) or SP1 (+79 to +87). To examine which PRRSV protein is responsible for TFDP2 induction, 3D4/21 cells were co-transfected with the plasmid that encodes PRRSV non-structural and structural protein (nsp1α, nsp1β, nsp4, nsp5, nsp7, nsp9, nsp10, nsp11, and N) or empty vector, −17/400-Luc, and the pRL-TK Renilla luciferase reporter plasmid. The results show that PRRSV N protein induced the highest TFDP2 promoter activity (Fig. 3D) and in a dose-dependent manner (Fig. 3E).
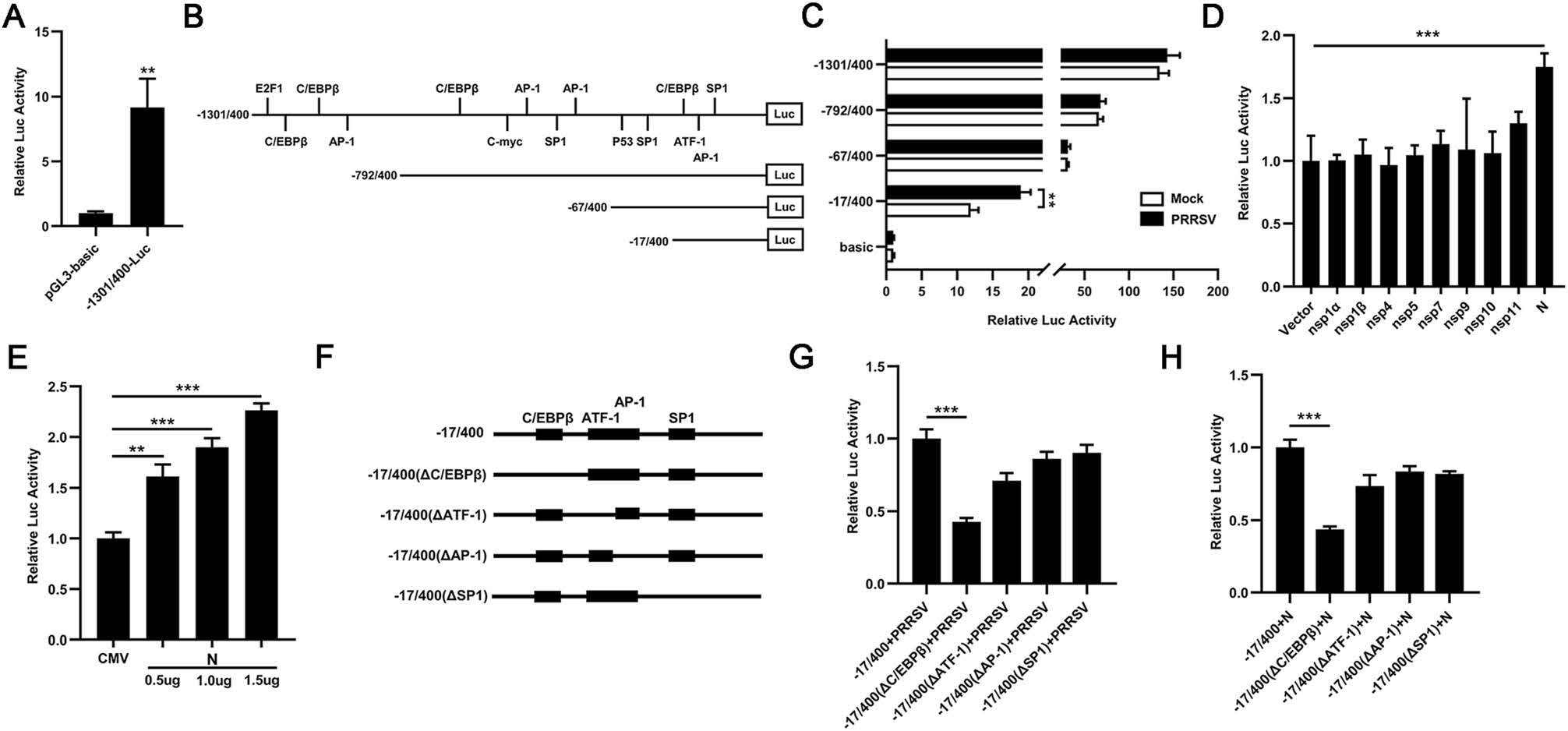
Figure 3. TFDP2 promoter was activated by PRRSV and its N protein. A, B 1701 bp TFDP2 promoter was cloned into pGL3-basic vector (A). The positions of the putative transcriptional regulatory elements in TFDP2 gene 5′ flanking region were identified and the promoter truncated mutants were generated (B). C The TFDP2 promoter truncated mutants or pGL3-basic and the pRL-TK Renilla luciferase reporter plasmid were co-transfected into 3D4/21 cells. Twenty-four hours later, cells were mock-infected or infected with 0.5 MOI PRRSV. At 24 h postinfection, cells were collected to determine luciferase activity. D 3D4/21 cells were co-transfected with a series of plasmids that encode PRRSV protein (nsp1α, nsp1β, nsp4, nsp5, nsp7, nsp9, nsp10, nsp11, and N) or empty vector, − 17/400-Luc, and the pRL-TK Renilla luciferase reporter plasmid. After 24 h, the cells were harvested for luciferase activity analysis. E 3D4/21 cells were co-transfected with Flag-N plasmid at doses of 0.5, 1.0, and 1.5 μg or empty vector, −17/400-Luc, and the pRL-TK Renilla luciferase reporter plasmid, and the cells were harvested for luciferase activity analysis at 24 h post-transfection. F Schematic representation of putative TFDP2 promoter binding sites between −17 to + 400 bp region, including the C/EBPβ (−11 to −4), ATF-1 (+30 to +40), AP-1 (+37 to +43) or SP1 (+79 to +87). G and H 3D4/21 cells were co-transfected with a series of TFDP2 promoter binding sites mutants and the pRL-TK Renilla luciferase reporter plasmid for 24 h and cells were then infected with 0.5 MOI PRRSV (G) or transfected with Flag-N plasmid (H). Twenty-four hours later, cells were harvested for luciferase activity analysis. Data are representative of three independent experiments. Significant differences are indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further identify the main regulatory element responsive to PRRSV, we mutated putative binding sites of C/EBPβ, ATF-1, AP-1 and SP1 from − 17/400-Luc, respectively (Fig. 3F). The results of luciferase assay demonstrated that mutation in the C/EBPβ binding site remarkably impaired PRRSV and N-induced activation of TFDP2 promoter (Fig. 3G and 3H). Therefore, these results indicate that the C/EBPβ binding site of TFDP2 promoter is the main responsive element upon PRRSV infection.
-
To further clarify the role of C/EBPβ in PRRSV and N-induced TFDP2 production, we detected the effect of knockdown of C/EBPβ on the TFDP2 expression induced by PRRSV and N protein using real-time RT-qPCR. The knockdown efficiencies of C/EBPβ siRNAs were identified by real-time RT-qPCR and Western blot (Fig. 4A). As is shown in Fig. 4B and 4C, silencing C/EBPβ significantly dampened PRRSV and N-induced up-regulation of TFDP2 expression. These results suggest the involvement of C/EBPβ in PRRSV and N-induced TFDP2 expression.
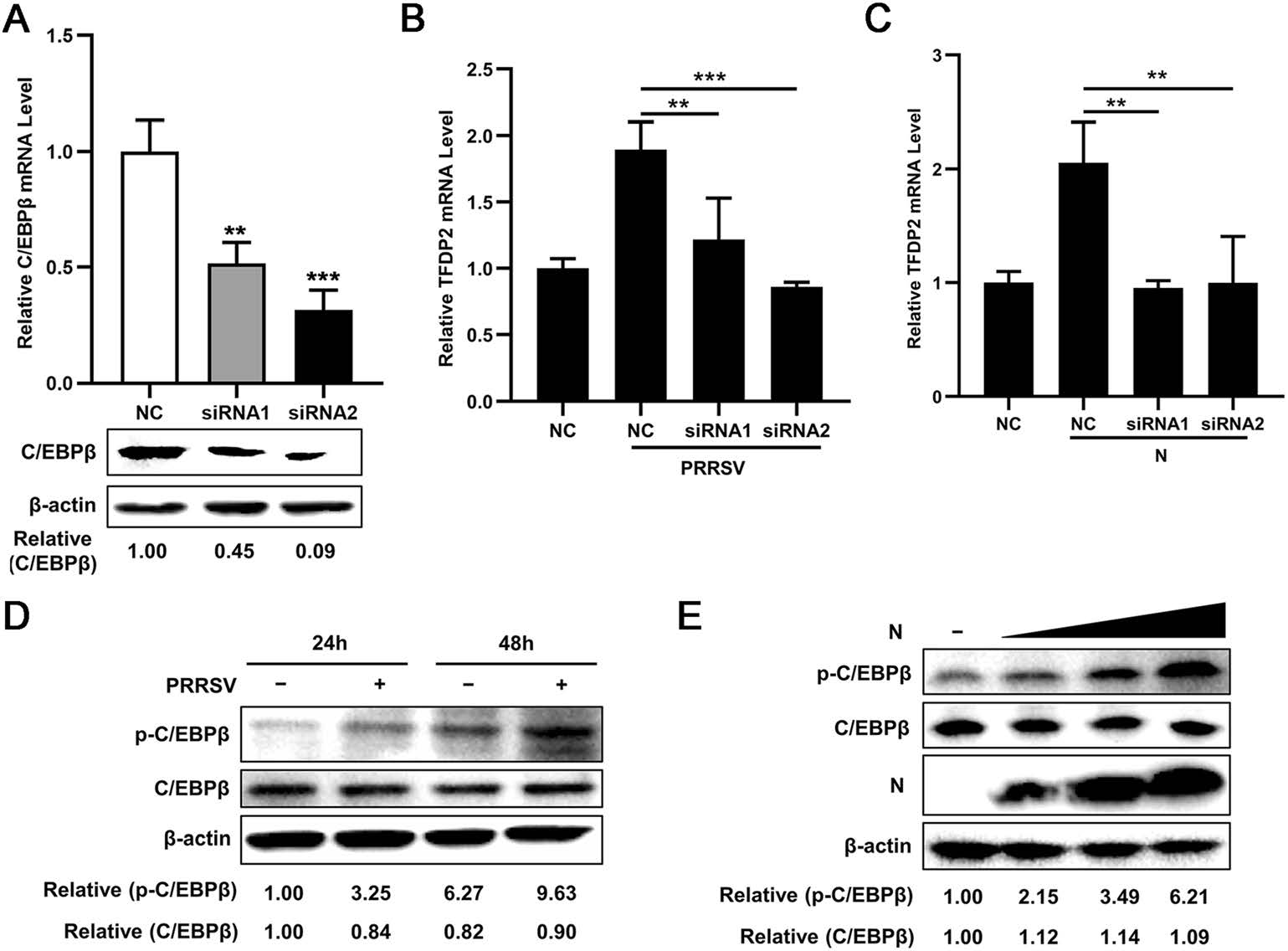
Figure 4. C/EBPβ is required for PRRSV and N-induced TFDP2 expression. A C/EBPβ siRNAs interference effects were detected by real-time RT-qPCR and Western blot. B, C 3D4/21 cells were transfected with C/EBPβ siRNAs, at 24 h post-transfection, cells were mock-infected or infected with PRRSV (B), or transfected with Flag-N plasmid or empty vector (C). After 24 h, total RNAs were extracted for TFDP2 mRNA analysis by real-time RT-qPCR. D 3D4/21 cells were inoculated with or without PRRSV (MOI = 0.5) for 24 h or 48 h, and cells were then harvested for p–C/EBPβ and C/EBPβ analysis by western blot. E 3D4/21 cells were transfected with Flag-N plasmid at doses of 0.5, 1.0, and 1.5 μg, an empty vector was used as a control, 24 h later, cells were collected for analysis of p–C/EBPβ and C/EBPβ levels using Western blotting. Data are representative of three independent experiments. Significant differences are indicated as follows: **, P < 0.01; ***, P < 0.001
Previous studies have shown that PRRSV can activate C/EBPβ by enhancing phosphorylation of C/EBPβ (Bi et al. 2014; Wang et al. 2019). To validate this finding, 3D4/21 cells were infected with or without PRRSV and harvested at 24 or 48 h postinfection (hpi), and the level of p–C/EBPβ and C/EBPβ were analyzed by Western blot. Consistent with previous studies, our results showed increased phosphorylation of C/EBPβ after PRRSV infection (Fig. 4D). Furthermore, we found that overexpression of PRRSV N protein increased p–C/EBPβ level in a dose-dependent manner (Fig. 4E). Collectively, these data demonstrate that both PRRSV and its N protein induce TFDP2 production by activating C/EBPβ.
-
It has been reported that cyclin A is the target gene of transcription factors E2F/DP complex, and it controls the S phase progression of the cell cycle (Yam et al. 2002). We, therefore, speculated whether TFDP2 affects S phase progression through regulating cyclin A production. First, we constructed the luciferase reporter plasmid of cyclin A and identified its promoter activity (Fig. 5A). Next, we overexpressed TFDP2 to figure out the effect of TFDP2 on cyclin A promoter. The results showed that TFDP2 overexpression induced the promoter activity of cyclin A (Fig. 5B). Real-time RT-qPCR analysis further confirmed that TFDP2 significantly increased the mRNA level of cyclin A (Fig. 5C). We then measured the cell cycle in TFDP2 overexpressed cells. As shown in Fig. 5D, overexpression of TFDP2 led to a decrease in the number of cells in the S phase. Meanwhile, we found that PRRSV infection up-regulated the expression of cyclin A, while knockdown of TFDP2 attenuated this up-regulation effect (Fig. 5E). Correspondingly, silencing TFDP2 restored the reduction effect of PRRSV on the number of cells in the S phase (Fig. 5F). Taken together, these results indicate that PRRSV infection accelerates the S phase progression of the cell cycle through TFDP2-mediated up-regulation of cyclin A expression.
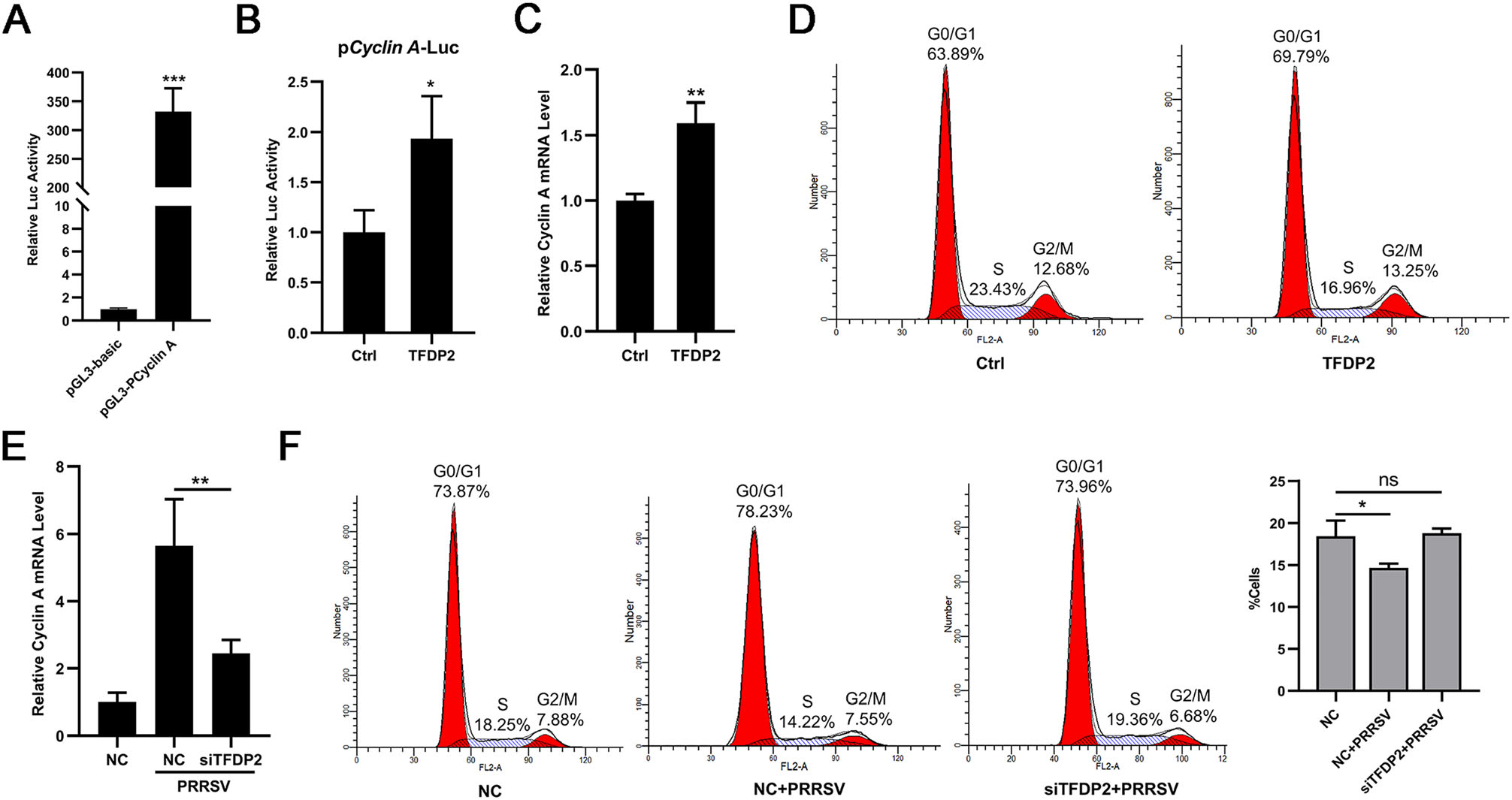
Figure 5. Effect of TFDP2 on the regulation of the cell cycle in 3D4/21 cells. A Identification of cyclin A promoter activity. 293 T cells were co-transfected with cyclin A promoter-reporter plasmid or pGL3-basic and the pRL-TK Renilla luciferase reporter plasmid for 24 h to detect luciferase activity. B 3D4/21 cells were co-transfected with cyclin A promoter plasmid, the pRL-TK Renilla luciferase reporter plasmid and Flag-TFDP2 or empty vectors. At 24 h post-transfection, cells were harvested for luciferase activity analysis. C and D 3D4/21 cells were co-transfected Flag-TFDP2 or empty vectors for 24 h, total RNAs were extracted for cyclin A mRNA analysis (C), and cells were stained with propidium iodide and the cell cycle distribution was analyzed by flow cytometry (D). E and F 3D4/21 cells were transfected with NC or siTFDP2 for 24 h and then mock-infected or infected with 0.5 MOI PRRSV for 24 h. Total RNAs were extracted for cyclin A mRNA analysis (E), and cells were stained with propidium iodide for cell cycle analysis by flow cytometry (F). Data are representative of three independent experiments. Significant differences are indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, no significant.
-
Accumulating studies have demonstrated that viruses can regulate the host cellular life cycle to favor their proliferation (Fan et al. 2018; Nascimento et al. 2012). To better understand the role of cell cycle on PRRSV proliferation, 3D4/21 cells were synchronized to G0/G1 phase, S phase, or G2/M phase, by cell starvation, thymidine, or nocodazole treatment, respectively. Flow cytometry analysis showed cell cycle stages were arrested successfully by serum starvation and drug treatment (Fig. 6A). First, we examined the relationship between the cell cycle and virus adsorption. PRRSV was inoculated in different cell cycles for 1 h, after which the cells were immediately collected to detect the PRRSV ORF7 level by real-time RT-qPCR. As shown in Fig. 6B, the cells in the S phase were not conducive to PRRSV adsorption.

Figure 6. Sensitivity to PRRSV infection in different cell cycles. A 3D4/21 cells were arrested in the G0/G1, S or G2/M phases of the cell cycle using serum starvation, thymidine (2 mmol/L), or Nocodazole (80 ng/mL), respectively. The effect of cell cycle synchronization was examined by flow cytometry. B 3D4/21 cells in different cell cycles were inoculated with PRRSV for 1 h at 4 ℃ and were immediately collected to detect PRRSV ORF7 level by real-time RT-qPCR. C−F 3D4/21 cells in different cell cycles were inoculated with 0.5 MOI PRRSV for 24 h. PRRSV ORF7 level was measured by real-time RT-qPCR (C) and PRRSV Nsp2 level was determined by immunofluorescence (scale bar: 20 μm) (D) and Western blotting (E). PRRSV titers in supernatants were tested by TCID50 assay (F). Data are representative of three independent experiments. Significant differences are indicated as follows: **, P < 0.01; ***, P < 0.001
To further assess the effects of synchronization at different cell cycle stages on PRRSV replication, PRRSV was inoculated in different cell cycles and the infected cells were harvested after 24 h. Real-time RT-qPCR analysis confirmed that PRRSV ORF7 level in the S phase was lower than that in the G2/M or G0/G1 phases (Fig. 6C). Besides, immunofluorescence showed that the expression level of PRRSV Nsp2 was lowest in the S phase (Fig. 6D) and the results of Western blotting further confirmed this finding (Fig. 6E). Furthermore, the virus titers in supernatants of cells in the G2/M or G0/G1 phases appeared greater than that in the S phase (Fig. 6F). The evidence presented indicates that PRRSV replication in 3D4/21 cells is sensitive to the G1/G0 or G2/M phases, rather than the S phase. Together, these results indicate that the cells in the S phase were not conducive to PRRSV adsorption and replication.
PRRSV Up-Regulates TFDP2 Levels in Viral Infected 3D4/21 Cells
TFDP2 Contributes to PRRSV Proliferation
C/EBPβ Response Element Is Indispensable for PRRSV and Its N Protein to Activate TFDP2 Promoter
C/EBPβ Is Critical for PRRSV and N-Induced TFDP2 Expression
PRRSV Regulates Cell Cycle at S Phase through TFDP2-Mediated Up-Regulation of Cyclin A
The Decrease of the S Phase Favors the Proliferation of PRRSV
-
PRRSV is a critical pathogen of swine that brings serious losses to pig farms (Neumann et al. 2005). TFDP2 dimerizes with E2F to regulate the transcription of a series of genes, including cell cycle progression, DNA repair, cell differentiation, and apoptosis (Attwooll et al. 2004; DeGregori and Johnson 2006). In this study, we found that the expression of TFDP2 was upregulated during PRRSV infection, and the underlying mechanism was investigated. We constructed the promoter-reporter plasmid of TFDP2 and its truncated mutants. The results of the luciferase assay demonstrated that PRRSV and N protein activated the promoter activity of TFDP2 to induce its production. We further found that C/EBPβ was essential for PRRSV or N-induced TFDP2 production. The phosphorylation of C/EBPβ was increased during PRRSV infection and knockdown of C/EBPβ significantly dampened PRRSV and N-induced expression of TFDP2. It has been reported that PRRSV infection can activate C/EBPβ (Bi et al. 2014; Wang et al. 2019), which is consistence with our findings.
Next, we investigated the effect of TFDP2 on PRRSV proliferation. Our results showed that overexpression of TFDP2 promoted the proliferation of PRRSV, and, correspondingly, TFDP2 knockdown suppressed PRRSV proliferation. Since TFDP2 is involved in regulating genes about the cell cycle, we speculated that weather PRRSV affected the cell cycle via TFDP2 to promote its proliferation.
Cyclin A plays an important role in controlling the S phase progression of the cell cycle and its promoter has been shown to be regulated by transcription factors E2F/DP (Yam et al. 2002). Chronic hepatitis C virus (HCV) NS2 protein induces the cell cycle arrest at the S phase through the down-regulation of cyclin A expression (Yang et al. 2006). PCV2 replication is both S and G2/M phase-dependent, and PCV2 infection down-regulates GPNMB expression, leading to the down-regulation of cyclin A and the S phase accumulation, which provides favorable conditions for efficient viral replication (Guo et al. 2019; Tang et al. 2013; Xu et al. 2016). In this study, we found that PRRSV infection decreased the number of cells in the S phase through TFDP2-induced upregulation of cyclin A expression.
PRRSV N protein has been suggested to regulate the cell cycle progression (Yoo et al. 2003). In contrast to our findings that PRRSV infection accelerates the S phase progression, another study demonstrated that PRRSV nsp11 causes the delay of MARC-145 cells at the S phase (Sun et al. 2014). Besides, it has been shown that PRRSV and its truncated glycoprotein 5 (GP5Δ97−119) can induce arrest of MARC-145 cells at the G2/M phase (Mu et al. 2015; Song et al. 2018), which were not observed in our study. These discrepancies may be due to the use of different cell lines or other experimental conditions. We speculate that the effects of PRRSV infection on the cell cycle may be cell type-specific.
It has been generally accepted that many viruses manipulate the cell cycle of infected cells to regulate and amplify their infection. Rotavirus induction of cell cycle arrest in the S/G2 phase enhances viral replication (Glück et al. 2017). Herpesviruses (Flemington 2001), severe acute respiratory syndrome coronavirus (SARS-CoV) 3b protein (Yuan et al. 2005), influenza A virus and its NS1 protein (He et al. 2010; Jiang et al. 2013) and murine norovirus (MNV) (Davies et al. 2015) can induce cell cycle arrest in the G0/G1 phase. Enterovirus 71 (EV71) (Yu et al. 2015) and Porcine epidemic diarrhea virus (PEDV) M protein (Xu et al. 2015) mediates cell cycle arrest in the S phase. We found that synchronization cells in the G0/G1 or G2/M phase rather than the S phase are more favorable to PRRSV proliferation.
Thus, we believe that PRRSV infection decreases the number of cells in the S phase is beneficial for its proliferation. One possibility is that the S phase may provide fewer ribonucleotides for PRRSV RNA synthesis than the G0/G1 phase. Since ribonucleotides are precursors of deoxyribonucleotides, and cells in the S phase will reduce the levels of ribonucleotides (Chen and Makino 2004). Another possibility is that the G2/M phase helps to establish a pseudo-S phase state, which may be more conducive to virus replication (Davy and Doorbar 2007).
In summary, our results showed that PRRSV infection upregulated TFDP2 production in 3D4/21cells by activating transcription factor C/EBPβ. Furthermore, our study demonstrated that PRRSV affected the cell cycle progression by regulating TFDP2 to favor its proliferation. However, the specific molecular mechanism still needs further investigation. In general, this study identified a novel mechanism by which PRRSV utilizes host proteins to promote its infection by affecting the cell cycle.
-
This work was supported by the National Key Research and Development Program of China (2018YFD0500500) and the National Natural Science Foundation of China (31272540).
-
JH conceived and designed the experiments. MZ, XL, RS, PS, AC, LZ performed the experiments. MZ, XL analyzed the data. MZ and JH wrote the paper.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.


 DownLoad:
DownLoad: