














HTML
-
Japanese encephalitis is a mosquito-borne zoonosis disease caused by Japanese encephalitis virus (JEV) (Lopez et al. 2015), which is mainly prevalent in Southeast Asia and maintains a zoonotic cycle between ardeidae birds or pigs and Culex pippens (van den Hurk et al. 2009; Impoinvil et al. 2013). JEV infects various animals and human beings (Impoinvil et al. 2013). It can develop into viral encephalitis, leading to permanent sequelae or death, especially in children (Solomon and Vaughn 2002; Campbell et al. 2011).
JEV belongs to the Flaviviridae family that is charac-terized by single, positive-stranded RNA viruses. Its gen-ome length is about 11 kb and encodes a large polyprotein. The polyprotein yields at least three structural proteins (capsid protein C, envelope protein E and precursor membrane protein prM) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5) upon cleavage by the virus and host's proteases (Unni et al. 2011; Morita et al. 2015). Flavivirus NS1 protein is involved in various biological events such as viral repli-cation and immune invasion during viral infection and transmission (Muller and Young 2013; Amorim et al. 2014; Rastogi et al. 2016). The Japanese encephalitis ser-ogroup of the neurogenic flavivirus utilizes programmed -1 ribosomal frameshifting to synthesize their NS1' protein. The sequence encoding NS2A is 3' adjacent to that encoding NS1. The occurrence of -1 PRF between codons 8 and 9 of sequence encoding NS2A adds 52 extra amino acids to the C-terminus of NS1 (Firth and Atkins 2009; Melian et al. 2010; Ye et al. 2012; Yun et al. 2016).
It is postulated that the NS1' protein synthesized by JEV and West Nile virus (WNV) through ribosomal frameshifting plays an important role in the neurological invasion of the virus, while the mechanism involved remains to be fully elucidated (Melian et al. 2010; Ye et al. 2012; Wang et al. 2015). It has been previously reported that NS1' co-localizes with NS1 and can substitute for NS1 during WNV replication (Young et al. 2013). Evolution-arily, WNV that expresses NS1' is more effective in virus transmission to mosquitoes (Melian et al. 2010). Frame-shift inactivation caused by pseudoknot mutation leads to lack of NS1' synthesis, thus forming a less virulent virus ideal for vaccination (Chambers et al. 2007; Sun et al. 2012). JEV NS2A G66A, the mutation used for this pur-pose, is a 3' pseudoknot of the frameshift site that serves as a recoding signal for the frameshifting. The frameshifting level is very low in the absence of the frameshift stimu-latory effect provided by the pseudoknot. Besides JEV, human immunodeficiency virus (HIV) also has a frameshift phenomenon, which requires strict control of the Gag-Pol to Gag ratio produced by -1 PRF to ensure efficient viral assembly, genome packaging and maturation (Karacostas et al. 1993; Hung et al. 1998; Shehu-Xhilaga et al. 2001; Dulude et al. 2006; Brakier-Gingras et al. 2012).
JEV NS3 protein is a multifunctional protein containing 619 amino acids, which possesses several enzymatic activities including serine protease activity, RNA helicase activity and nucleoside triphosphatase activity (Chambers et al. 1990; Preugschat et al. 1991; Li et al. 1999). The serine protease activity was identified within about one fourth of the N-terminal domain of the NS3 protein and the C-terminus of NS3 contains sequence motifs responsible for NTP binding, NTP hydrolysis and RNA helicase, accounting for three fourths of the total numbers of amino acids (Utama et al. 2000). Notably, NS3 protein is pro-posed as components of the RNA replicase and involved in JEV minus-strand RNA synthesis (Chen et al. 1997). Therefore, NS3 is considered to be one of the indispensable protein during JEV replication.
Activation of interferon (IFN) signal transduction path-way results in the up-regulation of hundreds of ISGs, such as C19orf66. This is also known as Shiftless, FLJ11286, UPF0515 or RyDEN (Suzuki et al. 2016; Balinsky et al. 2017; Wang et al. 2019). C19orf66 is in human chromo-some 19, open reading frame 66, encoding a 33.1 kDa protein-peptide containing 291 amino acids. The expres-sion of C19orf66 is regulated by type I and type II inter-feron (Singh et al. 2007; Taylor et al. 2008; Zimmerer et al. 2008; Schmeisser et al. 2010; Schoggins et al. 2011). Moreover, C19orf66 has antiviral activity against dengue virus (DENV) and encephalomyocarditis virus (EMCV), and can interact with DENV protein (Balinsky et al. 2017). Previous studies postulate that C19orf66 inhibits the replication of all DENV serotypes and it is a cellular mRNA binding protein that also interacts with MOV10, a RISC complex RNA helicase that is a member of the SF1 (Suzuki et al. 2016). MOV10 silencing results in an increased half-life of mRNA targets (Gregersen et al. 2014; Balinsky et al. 2017). Recent studies have identified that C19orf66 is an inhibitor of HIV programmed -1 ribosomal frameshifting (Wang et al. 2019). Cells infected with Kaposi's sarcoma-associated herpesvirus (KSHV) induce viral endoribonucleases to allow rapid decay of the host's RNA. Meanwhile, the 3' untranslated region (3' UTR) of the C19orf66 transcript protects them from the cleavage of the endoribonucleases (Rodriguez et al. 2019).
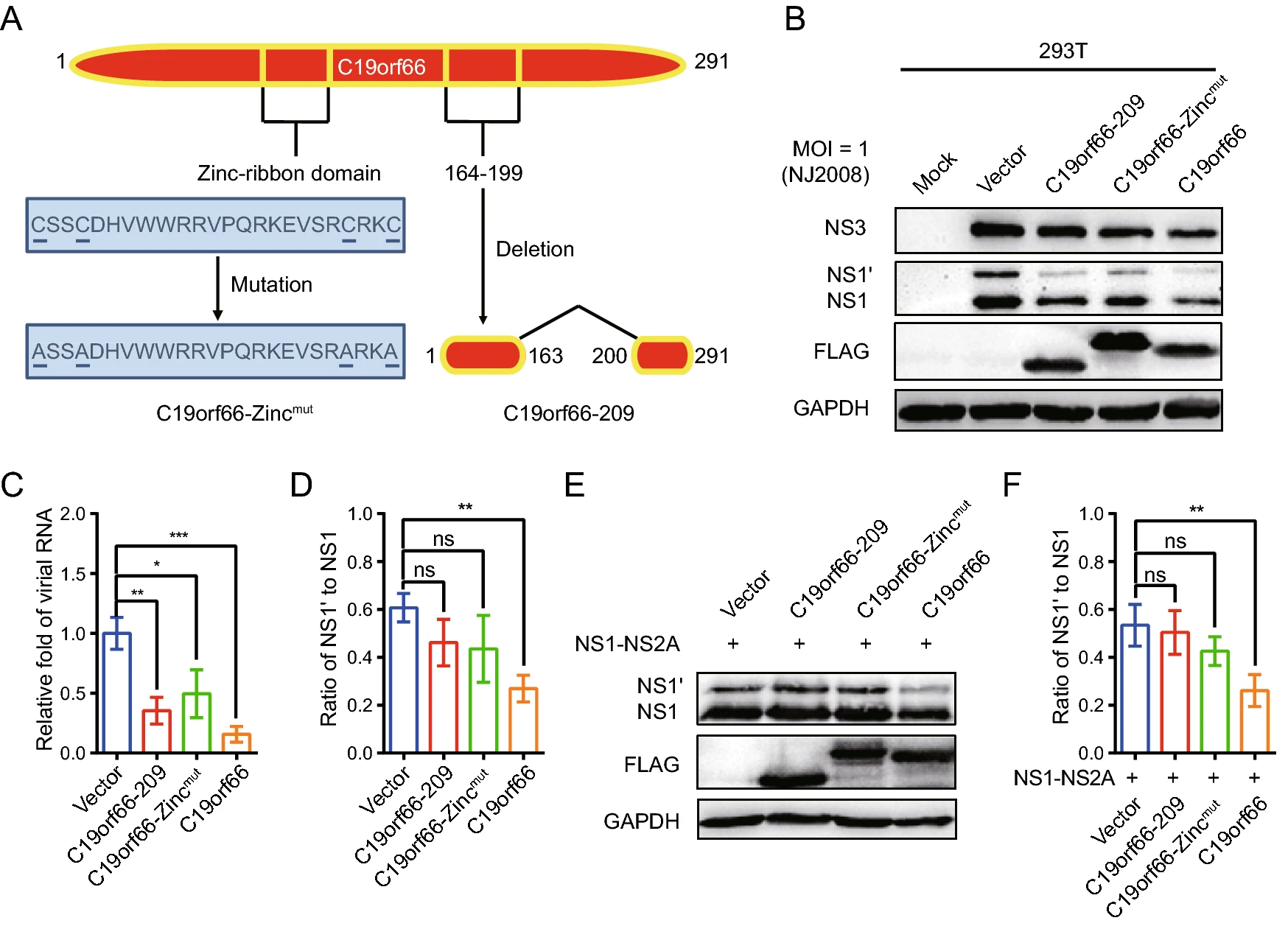
Alternative splicing generates two C19orf66 isoforms: the long and short form comprising 291 and 255 amino acids, respectively (Wang et al. 2019). It is suggested that the C19orf66 protein contains eight α-helixes, seven β-strands, nuclear localization signal (121–137), nuclear export signal (261–269), Zinc-ribbon domain (112–135), and coiled-coil motif (261–285). It also has a unique glu-tamic acid-rich (E-rich) domain in its C-terminus (Suzuki et al. 2016). Thirty-six amino acids (164–199) that form part of the fifth β-strand and the entire sixth β-strand are absent in the short form of C19orf66. C19orf66-209 cannot specifically bind to the -1 PRF signal for directly function in defense against HIV (Wang et al. 2019). Kinast et al. (2020) reported that C19orf66-209 and C19orf66-Zincmut restrict Hepatitis C virus (HCV) replication differently.
Herein, C19orf66 was identified as a novel ISG that inhibits JEV replication. Ectopic expression of C19orf66 inhibited JEV replication in 293T cells by hindering the frameshift-mediated expression of NS1'. However, it pro-moted the degradation of the JEV NS3 protein through the lysosome pathway. Meanwhile, C19orf66 knock-down cells had an increased viral titer. Moreover, C19orf66-209 and C19orf66-Zincmut cannot inhibit frameshift efficiency of JEV -1 PRF. Collectively, this study attests that C19orf66 antagonizes JEV infection differently, especially by targeting the NS1' and NS3 expression of JEV.
-
The C19orf66 polyclonal antibody ab122765 was sourced from Abcam, while the FLAG-tag polyclonal antibody D6W5B was sourced from GST. In the same line, the NS5 polyclonal antibody GTX131359 and NS3 polyclonal antibody GTX125868 were supplied by GeneTex (Irvine, CA, USA), while the GAPDH polyclonal antibody sc-25778 was sourced from SantaCruz (Dallas, TX, USA). However, monoclonal antibodies for JEV NS1 and NS1' were homemade. Inhibitors such as cycloheximide (CHX), MG-132, NH4Cl, 3-MA and Z-VAD-FMK were purchased from APExBIO (Houston, TX, USA).
-
Human interferon-stimulated genes such as OAS1, PKR, ISG15, C19orf66 and uncharacterized gene C19orf53 were cloned from A549 cells by RT-PCR. The cells were first stimulated using 104 IU/mL of IFN-α2b for 16 h, followed by total RNA extraction using Trizol (TaKaRa Bio, Kyoto, Japan) and the reverse transcription of the RNA to cDNA using the PrimeScriptTM II 1st Strand cDNA Synthesis Kit (TaKaRa). Specific primers were designed using the cDNA as templates, followed by PCR amplification and sequencing. FLAG tag encoded gene was added to the N-terminus of the target gene and subsequently inserted into pcDNA3.1(-) to generate eukaryotic expression vec-tors. The amino acid sequence of the C19orf66-Zincmut was mutated from -CXXC-16aa-CXXC- to -AXXA-16aa-AXXA- with X representing any amino acid (Suzuki et al. 2016; Balinsky et al. 2017; Wang et al. 2019). The RNA of the JEV strain NJ2008 was reverse-transcribed to cDNA using the aforementioned kit, followed by designing specific primers for different viral protein genes. Sixty-six bases at the 3' terminus of JEV E gene, the entire NS2A gene and a part of NS2B gene were combined to construct a plasmid capable of expressing JEV NS1 and NS1', named pNS1-NS2A (2412–4559 bp). Supplementary Table S1 lists the primers used for amplifying the various genes.
-
HEK-293T, HeLa and BHK-21 were grown in Dulbecco's modified essential medium (DMEM, GIBCO, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) (GIBCO), 100 U/mL penicillin and 100 μg/mL streptomycin, at 37 ℃ in 5% CO2. A549 cells and C6/36 cells were cultured in RPMI-1640 (GIBCO) supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin, at 37 ℃ and 28 ℃, respectively, in 5% CO2.
-
JEV strains SA14-14-2 (GenBank: JN604986.1), NJ2008 (GenBank: GQ918133), and HN2007 (GenBank: FJ495189.1) were maintained at - 80 ℃ in the laboratory and propagated in C6/36 cells. Virus titer levels were determined using the plaque-forming assay on BHK-21 cells. The BHK-21 cells were first seeded into a 6-well plate until they grew to 80%– 90%. The resultant virus-containing supernatant was dilu-ted tenfold and incubated at 37 ℃ for 2 h. The unbound virions in each well were then washed away by PBS, fol-lowed by the addition of DMEM containing 2% fetal bovine serum and 1% low melting agarose. The cell plates were maintained at 37 ℃ for three days upon coagulation, after which the virus titer was determined based on the number of plaques formed.
Infection with different JEV strains was carried out after 1 h of incubation at 37 ℃ and a specific multiplicity of infection (MOI). Unbound virions were washed away by PBS and cells were cultured for a specific period in DMEM (293T cells and HeLa cells) or RPMI-1640 (A549 cells) supplemented with 2% FBS, 100 U/mL penicillin and 100 U/mL streptomycin, at 37 ℃ in 5% CO2. The cell super-natants were then harvested for determination of the viral titer levels using the plaque-forming assay.
-
Plasmid transfection and co-transfection into viral cells were performed using Lipofectamine 3000 following the manufacturer's instructions. RNA interference was achieved by transferring siRNA at 50 nmol/L into the cells using the Lipofectamine RNAiMAX Reagent and subse-quent inoculation of JEV at specific MOI after 48 h of transfection. The siRNA targeting C19orf66 (sc-97810) and the control siRNA (sc-37007) were purchased from SantaCruz (Dallas, TX, USA).
-
The cells were lysed using radioimmunoprecipitation assay (RIPA) buffer for 15 min. The lysed cells were then cen-trifuged and the supernatant harvested as the protein sam-ples. The total protein concentration was determined using the BCA Protein Assay (Pierce), followed by the separation of 50 μg of the total protein from each sample by 10%-sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The resultant protein bands were then transferred onto a PVDF membrane (Bio-RAD Laborato-ries, Hercules, CA, USA). The membrane was then incu-bated with its corresponding primary antibody for 8 h at 4 ℃ after blocking with 5% nonfat milk. The membrane was washed thrice with PBST to remove the excess pri-mary antibody and subsequently incubated with the goat anti-mouse or goat anti-rabbit horseradish peroxidase (HRP) conjugated IgG antibody (Pierce) at 37 ℃ for 1 h. The protein banding patterns were then detected using the BIO-RAD Clarity Western ECL Substrate. The results obtained by Western blotting were analyzed with ImageJ software, and Grayscale analysis and quantification were performed according to the depth of the protein bands to calculate the protein ratio. One representative experiment out of three is shown.
-
Total RNA from JEV-infected cells was extracted using the OMEGA total RNA extraction kit (Bio-Tek, Norcross, GA, USA), followed by cDNA synthesis using the HiScript ® II 1st Strand cDNA Synthesis Kit (+ gDNA wiper) (Vazyme, Nanjing, China). The JEV RNA levels were then deter-mined by qRT-PCR using specific primers and a TB Green Premix Ex Taq (TliRNaseH Plus) (Takara) on a 7300 real-time PCR system (Applied Biosystems, Foster City, Cali-fornia, USA). The same procedure was used to extract total RNA from plasmid-transfected cells and detect their cor-responding RNA content. Supplementary Table S1 con-tains the details of all primers used.
-
Graphical and statistical analyses were performed using the GraphPad Prism 6.0 software (GraphPad Software Inc., La Jolla, CA, USA), and data presented as the mean of trip-licate experiments ± the standard deviation. The P value was calculated using the two-tailed Student's t-test at a significance threshold of 95%.
Antibodies
Plasmid Construction
Cell Line and Cell Culture
Virus Amplification, Inoculation and Titration
Plasmid Transfection and RNA Interference
Western Blot Analysis
Real-Time Polymerase Chain Reaction (PCR)
Statistical Analysis
-
Herein, several ISG reported having inhibitory effects on Flavivirus such as PKR, OAS1, ISG15, C19orf66 and uncharacterized gene C19orf53 were cloned and used to construct eukaryotic expression vectors followed by inoc-ulation with the JEV NJ2008 (MOI of 1) strain at 24 h post-transfection. C19orf66 had a significant inhibitory effect on JEV (Fig. 1A) and the virus titer in the cell supernatant of C19orf66 decreased by approximately 30-fold (Fig. 1B). Treatment of 293T, HeLa and A549 cells with recombinant human interferon IFN-α2b at 104 IU/mL result in increased expression of C19orf66 to varying degrees (Fig. 1C). Among the cells, the C19orf66 protein was significantly increased in HeLa and A549 cells. The qRT-PCR assays performed to detect the level of C19orf66 mRNA revealed a 121-fold, 11-fold and ninefold increase in the 293T, HeLa and A549 cells, respectively (Fig. 1D). Notably, the endogenous C19orf66 mRNA level in the 293T cells was very low. Though it increased sig-nificantly upon IFN-α2b stimulation, its level was still lower than in untreated HeLa and A549 cells. The expression of C19orf66 in A549 and HeLa cells upon IFN-α2b stimulation increased in a dose-dependent manner (Fig. 1E, 1F). Moreover, the level of C19orf66 protein in the JEV infected cells was higher than that of the control cells (Fig. 1G). The abundance of C19orf66 mRNA increased by approximately twofold, fourfold and fivefold in those cells respectively (Fig. 1H). These results indi-cated that C19orf66, a product of ISG, had an inhibitory effect on JEV. Its expression level increased after JEV infection.
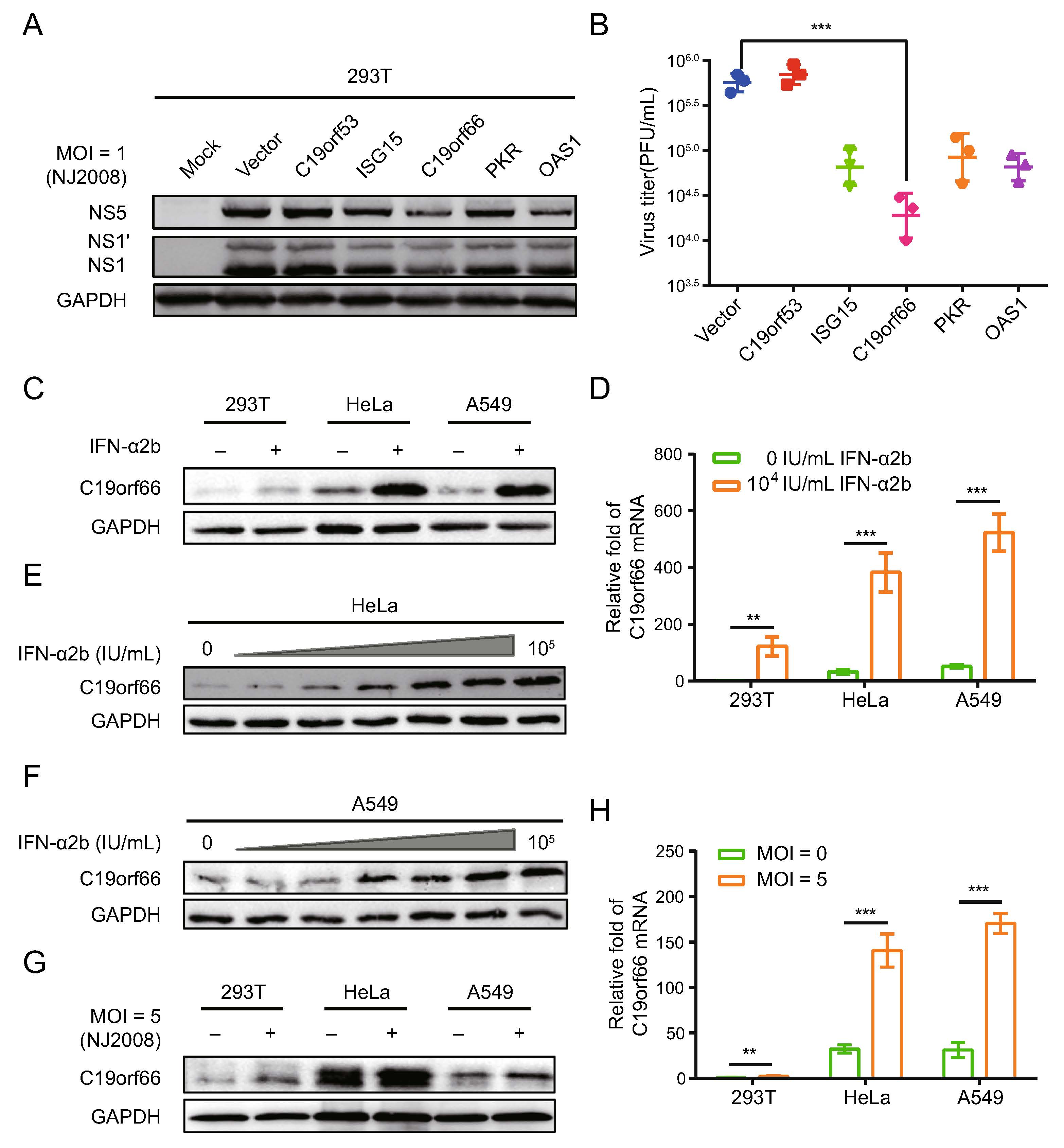
Figure 1. C19orf66 is an interferon stimulated gene product with significant anti-JEV activity. A Various ISGs were overexpressed in 293T cells. Cell lysates were harvested to detect the level of JEV NS1, NS1' and NS5 protein via Western blot analysis. B Supernatants were harvested and the production of progeny virions was determined by a plaque assay on BHK-21 cells. 293T, HeLa and A549 cells were treated with IFN-α2b at a concentration of 0 IU/mL or 104 IU/mL for 24 h, and the endogenous expression of C19orf66 and C19orf66 mRNA levels were detected by (C) Western blot analysis and (D) qRT-PCR, and normalized to the expression of GAPDH in each sample. E, F Lysates from HeLa and A549 cells treated with series concentrations of IFN-α2b (0, 1, 10, 102, 103, 104 and 105 IU/mL) for 24 h were harvested to analyze the endogenous expression of C19orf66 via Western blot analysis. Cells were challenged with JEV NJ2008 strain (MOI = 5) for 24 h, (G) Western blot analysis and (H) qRT-PCR were used to analyze the endogenous expression of C19orf66, and normalized to the expression of GAPDH in each sample. The data were expressed as the means ± SDs from three repeat experiments using t-test. **P < 0.01, ***P < 0.001.
-
The 293T cells were transfected with an expression vector encoding C19orf66 with an N-terminal Flag tag fusion or a negative control vector to determine if C19orf66 directly influences JEV replication. C19orf66 was poorly expressed in the 293T cell line, making it ideal for ectopic expression experiments. The cells were infected with the JEV NJ2008 strain at MOI of 0.1, 1 and 10 and harvested at 24, 36 and 48 h post-infection. The plaque-forming assays employed to determine the virus titer levels revealed a significant decrease in C19orf66 ectopically in the 293T cells than the negative control group, especially at 24 and 36 h post-in-fection. Compared to the control group, the decrease was 0.06-fold, 0.08-fold and 0.33-fold at 0.1, 1 and 10 MOI, respectively, at 24 h post-infection. The trend remained the same at 36 h post-infection (Fig. 2A). These results indi-cated that C19orf66 plays an inhibitory role in the early stages of JEV infection in a dose-dependent manner. Notably, the inhibitory effect of C19orf66 on JEV was not strain-specific because it exhibited a significant inhibitory effect on the replication of JEV HN2007, JEV SA14-14–2 and JEV SA14-14-2A66G strains in the 293T cells (Fig. 2B, 2C).
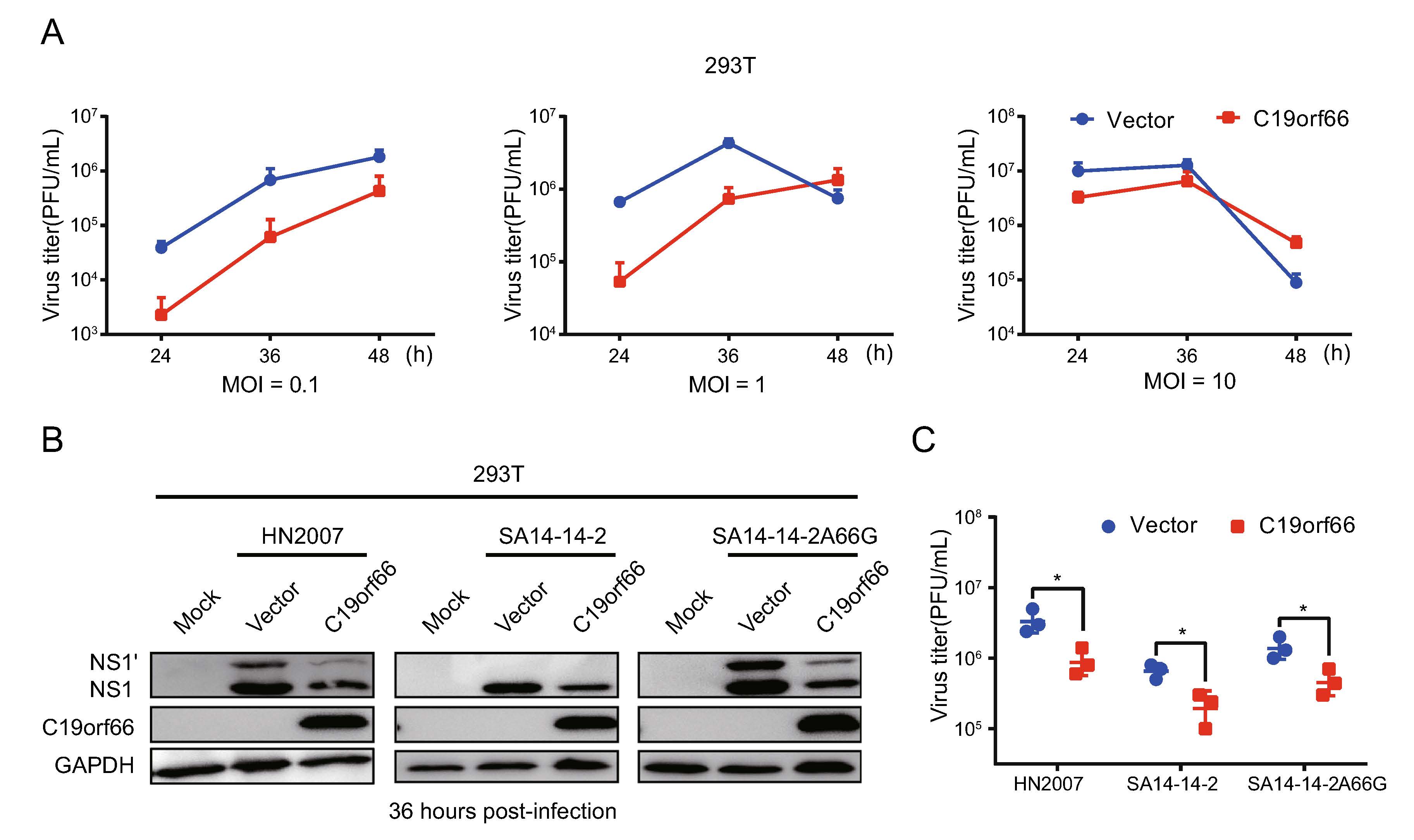
Figure 2. Ectopical expression of C19orf66 in 293T cells inhibits JEV infection. 293T cells were transfected with a plasmid encoding C19orf66 (0.5 μg) or an empty vector (0.5 μg) for 24 h and cells were challenged with JEV NJ2008 (MOI = 0.1, 1 or 10). A Supernatants were harvested at 24, 36 and 48 h post-infection, then the production of JEV virions were determined by a plaque assay titrated on BHK-21 cells. 293T cells transfected with a plasmid encoding C19orf66 (0.5 μg) or an empty vector (0.5 μg) were challenged with different strains of JEV (HN2007, NJ2008 or SA14-14-2A66G) at a 2 MOI. Cell lysates and supernatants were harvested at 36 h post-infection. B JEV infection was determined via the levels of NS1 and NS1' using Western blot analysis and (C) the production of JEV virions was determined by a plaque assay on BHK-21 cells. The data were expressed as the means ± SDs from three repeat experiments using t-test. *P < 0.05.
-
HeLa and A549 cells were treated with a small interfering RNA (siRNA) targeting C19orf66 or with a scramble siRNA to determine the involvement of endogenous C19orf66 in regulating viral replication. The cells were inoculated with JEV with varying MOI at 48 h after siRNA transfection. C19orf66 silencing led to increased virus titer in all MOI at 24 h post-infection (Fig. 3A, 3B). However, the increase was more significant at an MOI of 0.1 and 1 than that at MOI of 10. The virus titer in the supernatant collected at 12 and 24 h post-infection from the C19orf66 knock-down group was about ten times than that of the control. The proliferation of the three JEV strains after C19orf66 silencing in HeLa cells revealed that both the virus protein in the cell lysates and virus titer in the supernatant significantly increased at 36 h post-infection (Fig. 3C, 3D). These results demonstrated that endoge-nously expressed C19orf66 had a significant influence on JEV replication.
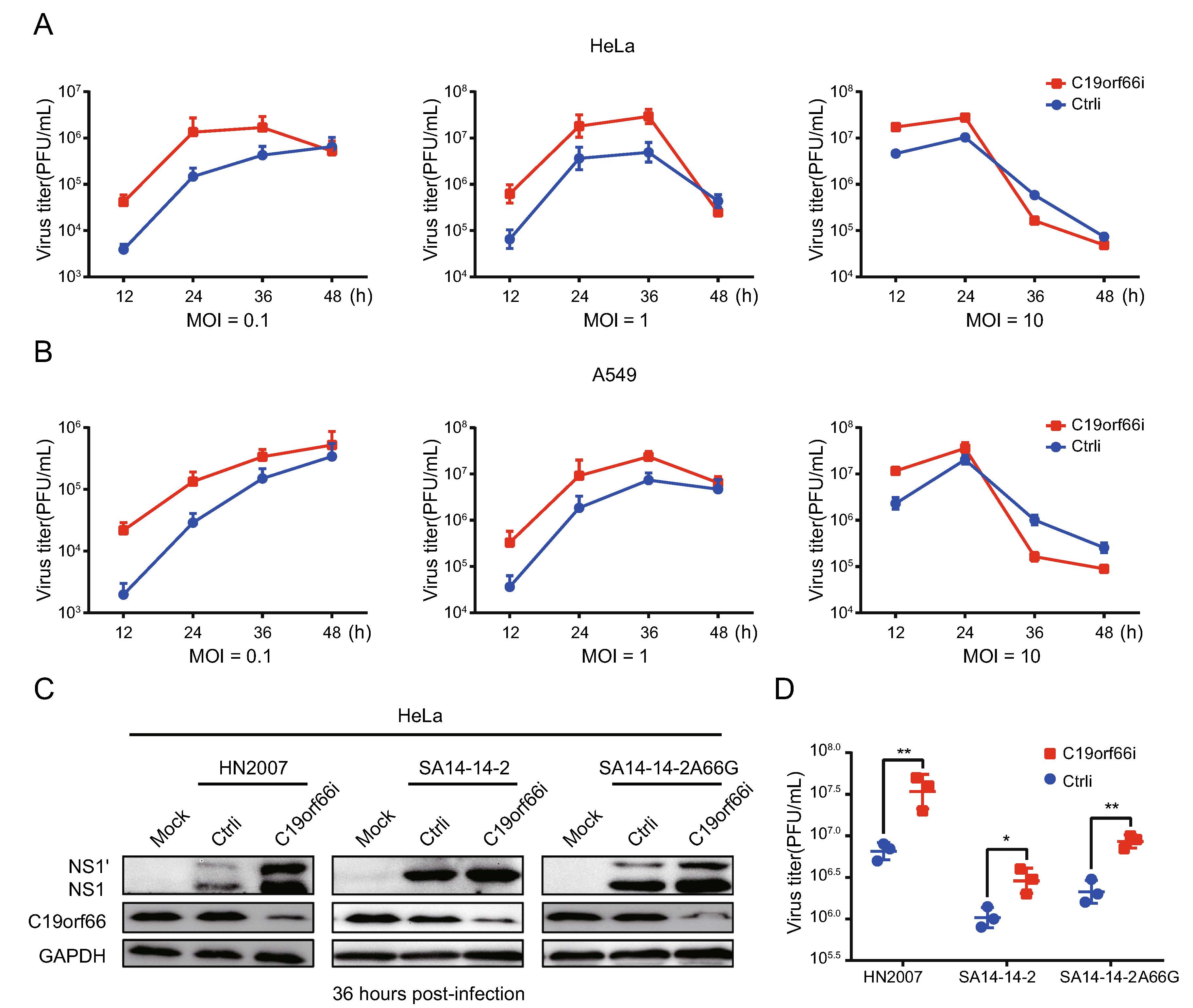
Figure 3. Knock-down of C19orf66 in HeLa and A549 cells leads to increased JEV production. A, B HeLa or A549 cells were transfected with C19orf66-specific siRNA or control siRNA using lipofectamine RNAiMAX Reagent. At 48 h post-transfection, cells were infected with JEV NJ2008 strain (MOI = 0.1, 1 or 10). At 12, 24, 36 and 48 h post-infection, supernatants were collected to detect the production of progeny virions by plaque assay. C HeLa cells transfected with C19orf66-specific siRNA or control siRNA were inoculated with different strains of JEV (HN2007, NJ2008 and SA14-14-2A66G) at MOI of 2 for 36 h, and cell lysates were harvested to detect the expression of NS1' and NS1 using Western blot analysis. D Super-natants were collected and the production of JEV virions was determined by a plaque assay. The data were expressed as the means ± SDs from three repeat experiments using t-test. *P < 0.05, **P < 0.01.
-
The ectopic expression of C19orf66 in 293T cells inhibited JEV replication (Fig. 4A) while silencing the expression of endogenous C19orf66 impaired the host's ability to resist the virus (Fig. 4B, 4C). Notably, compared to the control cells, the ratios of NS1' to NS1 in C19orf66 overexpression cells decreased by 55% (MOI of 0.1), 43% (MOI of 1) and 45% (MOI of 10) at 24 h post-infection (Fig. 4D). The ratio trend was the same at 36 and 48 h post-infection (Supplementary Fig. S1A–S1D). However, the ratios of NS1' to NS1 in JEV inoculated HeLa cells at 0.1, 1 and 10 MOI increased by 87%, 55% and 24%, respectively, after C19orf66 silencing (Fig. 4E), the trend in A549 cells was similar to that in HeLa cells (Fig. 4F). In the same line, the ratio of NS1' to NS1 in C19orf66 overexpressing cells decreased by 48% in the JEV HN2007 strain and 47% in the JEV SA14-14-2A66G strain at 36 h post-infection (MOI = 2) compared to the control cells (Supplementary Fig. S1E). However, the NS1' to NS1 ratio in C19orf66 knock-down cells increased by 40% in the JEV HN2007 strain and 63% in the JEV SA14-14-2A66G strain (Sup-plementary Fig. S1F). These findings strongly suggested that C19orf66 have a special effect against the JEV fra-meshift expression of NS1'.
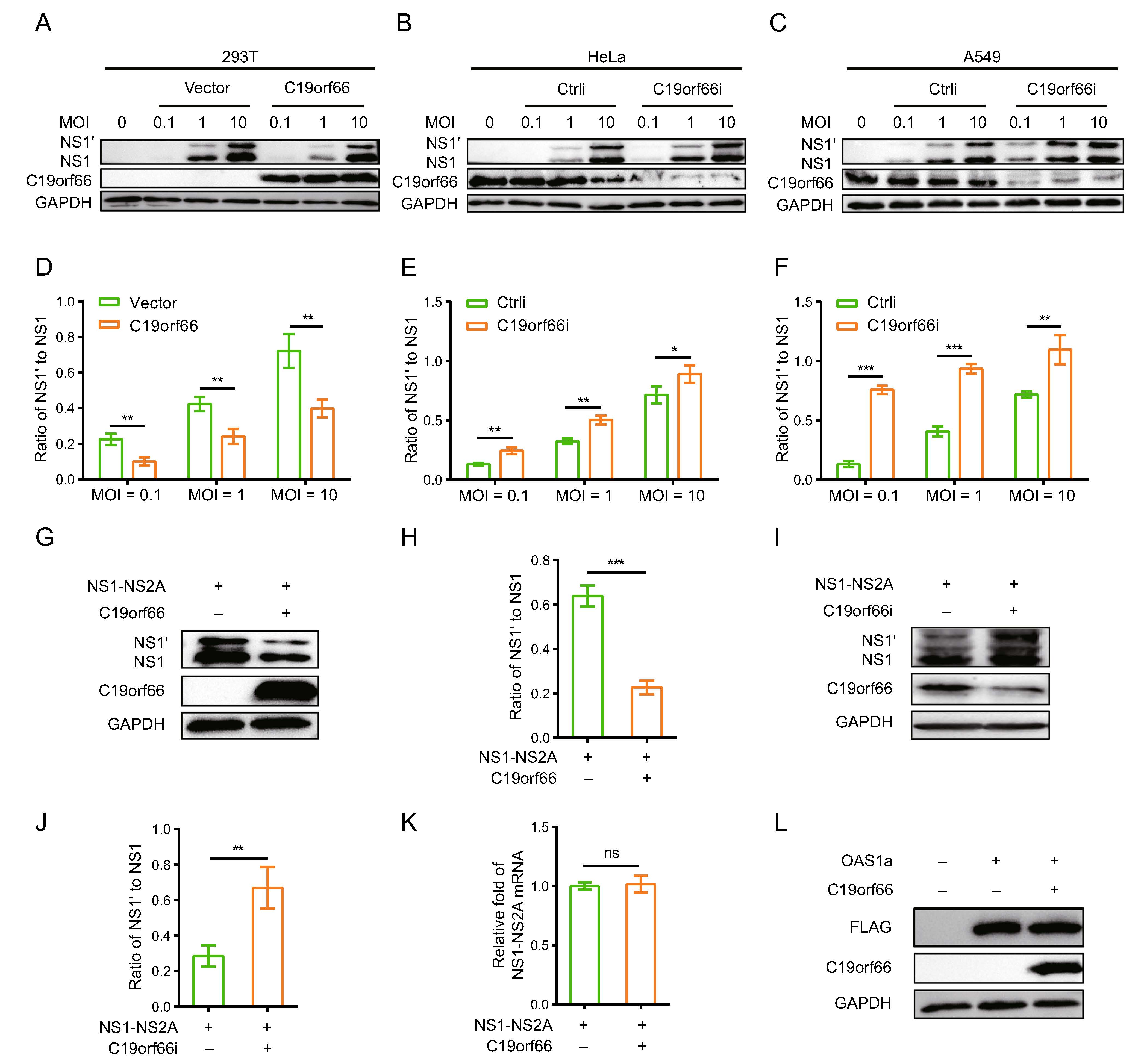
Figure 4. C19orf66 inhibits JEV -1 PRF and leads to a decrease in the ratio of NS1' to NS1. A, B 293T cells were transfected with a plasmid encoding C19orf66 (0.5 μg) or empty vector (0.5 μg) was used as a control, 24 h later, cells were inoculated with JEV NJ2008 strain (MOI = 0.1, 1 or 10) and cell lysates were harvested for Western blot analysis at 24 h post-infection. C–F HeLa and A549 cells were seeded in the 12-well plate, and then were transfected with C19orf66-specific siRNA or control siRNA, at 48 h post transfection cells were infected with JEV NJ2008 strain (MOI of 0.1, 1 or 10) for 24 h. Cell lysates were harvested to detect NS1' and NS1 by Western blot analysis with NS1-specific antibody. The gray values of NS1' and NS1 were quantified by ImageJ, and then the ratios of NS1' to NS1 were calculated. G Lysates from 293T cells co-transfected with NS1-NS2A plasmid (0.5 μg) and C19orf66 plasmid (0.5 μg) or vector plasmid (0.5 μg) for 24 h were collected to analyze the expression levels of NS1' and NS1 via Western blot analysis, and (H) the gray value of NS1' and NS1 were analyzed using ImageJ, then the ratios were calculated. I A549 cells were transfected with C19orf66-specific siRNA or control siRNA, and 36 h later, cells were transfected with a plasmid encoding NS1-NS2A (1 μg) for 24 h, then cell lysates were harvested to detect NS1' and NS1 levels through Western blot analysis with NS1-specific antibody. J The gray value of NS1' and NS1 were analyzed using ImageJ, then the ratios were calculated. K Quantitative real time PCR was used to analyze NS1-NS2A mRNA level in 293T cells 24 h after co-transfection with NS1-NS2A plasmid (0.5 μg) and with C19orf66 plasmid (0.5 μg) or control plasmid (0.5 μg). L Lysates from 293T cells co-transfected with a plasmid encoding OAS1a (0.5 μg) and a plasmid encoding C19orf66 (0.5 μg) for 24 h were collected for Western blot analysis using FLAG pAb. The data were expressed as the means ± SDs from three repeat experiments using t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
JEV NS1' protein is expressed via -1 PRF between the codons 8 and 9 of the sequence encoding NS2A. Consid-ering that C19orf66 may affect multiple aspects of virus propagation, co-transfecting pNS1-NS2A and pC19orf66 into 293T cells was used to analyze the function of C19orf66. The expression of NS1' and NS1 proteins and the ratio of NS1' to NS1 reduced in the C19orf66 over-expressed 293T cells (Fig. 4G, 4H). Conversely, C19orf66 silencing increased in the expression of NS1' or NS1 and the ratio of NS1' to NS1 (Fig. 4I, 4J). Meanwhile, qRT-PCR analysis revealed that cells co-transfected with pNS1-NS2A and pC19orf66 had a similar level of NS1-NS2A mRNA with cells co-transfected with pNS1-NS2A and pcDNA3.1(-). C19orf66 reduced the expression of NS1 and NS1' proteins without affecting mRNA transcription (Fig. 4K).
Given that C19orf66 has the possibility of non-specifi-cally inducing another protein degradation, 293T cells co-transfected with pOAS1a and pC19orf66 were used to analyze this possibility. Porcine OAS1, a member of the porcine OAS family, encodes two splice variants (OAS1a and OAS1b) that share 89.1% amino acid sequence identity (Perelygin et al. 2006; Zheng et al. 2016). Moreover, porcine OAS1 is 73% identical in sequence to human OAS1 and has similar enzymatic characteristics (Hartmann et al. 2003). Co-expression of OAS1a and C19orf66 in the 293T cells did not significantly affect the expression level of OAS1a (Fig. 4L), which clarifies that C19orf66 has specificity in reducing NS1' expression.
-
The crucial protein domains of C19orf66 in JEV antiviral activity, especially for its -1 PRF inhibition activity, were determined. The determination was done cognizant that a Zinc-finger motif present in the long but not in the short form of C19orf66 is required for the long form's role in inhibiting HCV (Kinast et al. 2020). C19orf66-209, C19orf66-Zincmut and C19orf66 were overexpressed in 293T cells followed by inoculation of the JEV NJ2008 strain into the cells to evaluate the antiviral role of the three proteins (Fig. 5A). Notably, C19orf66-209 and C19orf66-showed antiviral effects but were weaker than C19orf66 (Fig. 5B, 5C). JEV replication in the 293T cells transiently expressing C19orf66-209, C19orf66-Zincmut and C19orf66 was approximately 0.35-fold, 0.50-fold and 0.16-fold, respectively, compared to the control cells transfected with an empty vector (Fig. 5D). However, C19orf66-209 and C19orf66-Zincmut did not significantly reduce the NS1' to NS1 ratio (Fig. 5E). They also did not significantly change the expression and ratio of NS1' to NS1 when co-transfected with pNS1-NS2A into 293T cells (Fig. 5F). These findings indicated that the Zinc-finger domain and 164–199 amino acids are important domains for C19orf66 to exert frameshift inhibition function.
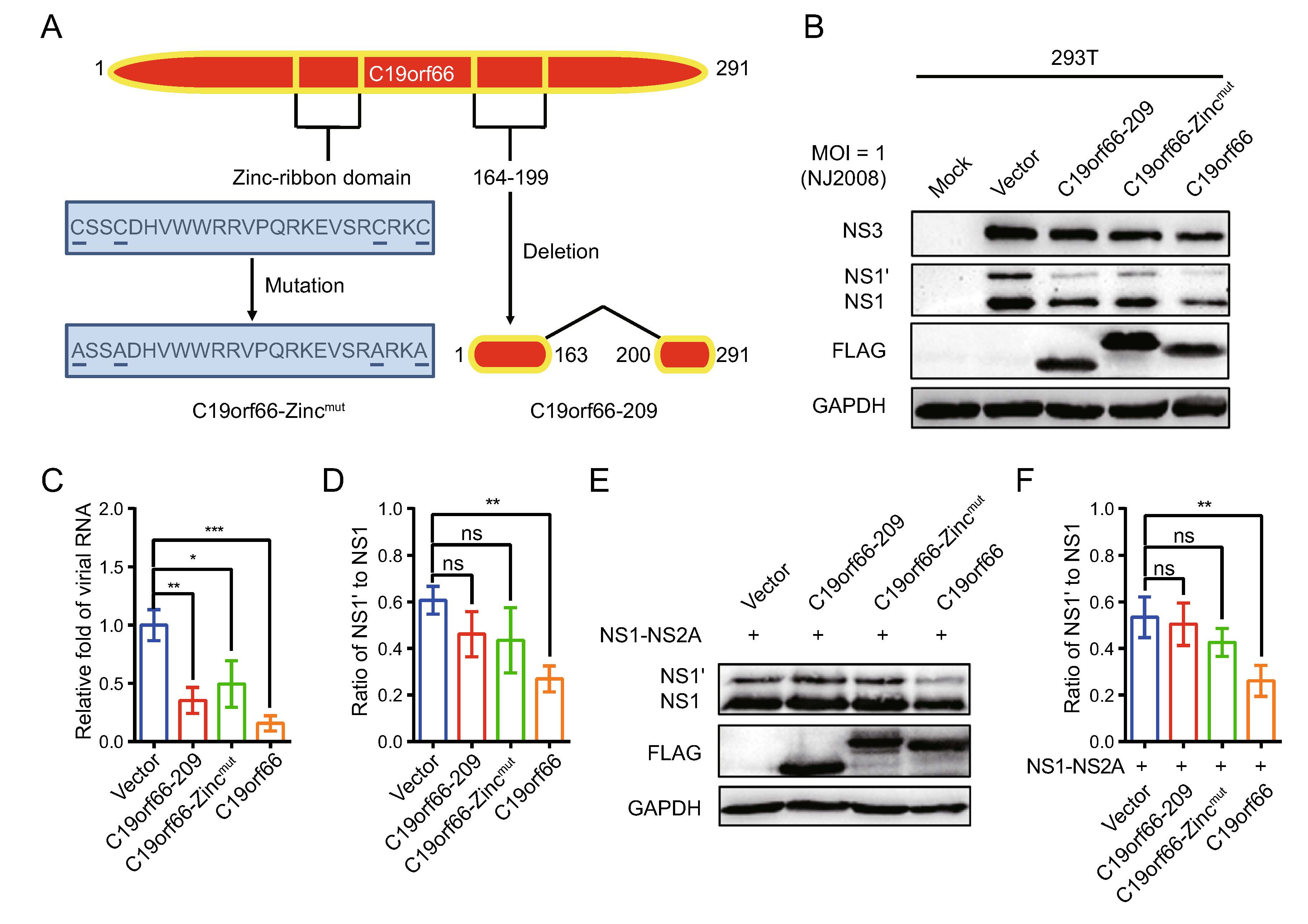
Figure 5. The Zinc-finger motif of C19orf66 is crucial for its restriction capacity on JEV -1 PRF. A C19orf66 protein (291 amino acids) contains NLS (121–137), NES (261–269), Zinc-ribbon domain (112–135), and coiled-coil motif (261–285). A unique glutamic acid-rich (E-rich) domain was also found in the C-terminus. B 293T cells transfected with plasmids encoding C19orf66-209 (1 μg), C19orf66-Zincmut (1 μg), C19orf66 (1 μg) or empty vector (1 μg) were inoculated with JEV NJ2008 strain at a MOI of 1, and 24 h later, cell lysates were harvested to detect the expression levels of JEV NS1, NS1' and NS3 via Western blot analysis. GAPDH was used as an internal control. C The supernatants were collected for qRT-PCR analysis and normalized to GPADH mRNA. D The ratios of NS1' to NS1 in different treatment groups were calculated. E 293T cells were co-transfected with plasmids encoding NS1-NS2A (0.4 μg) and C19orf66-209 (0.6 μg), C19orf66-Zincmut (0.6 μg), C19orf66 (0.6 μg) or vector plasmid (0.6 μg) for 24 h. The expression levels of C19orf66, NS1', NS1 and GAPDH were detected by Western blotting. F The ratios of NS1' to NS1 were calculated after analyzing the gray value of NS1' and NS1. The data were expressed as the means ± SDs from three repeat experiments using t-test. *P < 0.05, **P < 0.01, ***P < 0.001.
-
The abundance of the NS3 protein is high in JEV infected cells and it has multiple functions in JEV's life cycle. Notably, the expression of the NS3 protein in 293T cells ectopically expressing C19orf66 during JEV infection was significantly lower than that of 293T cells ectopically expressing C19orf66-209 or C19orf66-Zincmut (Fig. 5B). C19orf66 down-regulates the JEV NS3 protein expression by directly targeting the NS3 protein or influencing the viral replication. Herein, an NS3 expression vector was constructed and co-transfected with pC19orf66-209, pC19orf66-Zincmut or pC19orf66 into 293T cells to explore the direct influence of C19orf66 on the NS3 protein. There was a significant decline in the expression of the NS3 protein in cells co-expressing C19orf66 compared to those in the empty vector control group. Its expression was also not significantly changed after co-expression with C19orf66-209 or C19orf66-Zincmut (Fig. 6A). When a series of pC19orf66 doses and pNS3 were co-transfected into 293T cells, the expression of the NS3 protein decreased linearly with the increase in pC19orf66 dosage (Fig. 6B). The reduction in the expression of the NS3 protein caused by C19orf66 may be attributed to the down-regulation of NS3 mRNA. The abundance of NS3 mRNA in differently treated cells was thus measured using qRT-PCR to verify this possibility. Notably, overexpression of C19orf66, C19orf66-209 or C19orf66-Zincmut did not affect NS3 mRNA abundance, suggesting that the inhibit-ing effect of C19orf66 on NS3 protein expression was not caused by a decline in NS3 mRNA abundance (Fig. 6C). The mechanism by which C19orf66 degrades the NS3 protein was further investigated by co-transfecting pNS3 into 293T cells with pC19orf66 or control plasmid for 24 h followed by cycloheximide treatment of the cells to inhibit protein synthesis. C19orf66 overexpression accelerated the degradation of the NS3 protein (Fig. 6D). Linear regression analysis further revealed that the half-life (t1/2) of NS3 in C19orf66 overexpressed cells and the control group was 13.7 and 20.9 h, respectively (Fig. 6E). The C19orf66-mediated NS3 degradation pathway was further explored by co-transfecting 293T cells with pC19orf66 and pNS3 for 24 h and subsequently treating the cells with either the lysosomal acidification inhibitor, NH4Cl, the ubiquitin– proteasome inhibitor, MG-132, the autophagosome-for-mation inhibitor, 3-MA or the pan-caspase inhibitor, Z-VAD-FMK. NH4Cl treatment significantly reversed C19orf66-induced NS3 degradation, while 3-MA and Z-VAD-FMK did not affect NS3 degradation (Fig. 6F, 6G). Notably, MG-132 caused an increase in the NS3 protein compared to the control cells regardless of whether C19orf66 was overexpressed or not. This phenomenon suggested that the ubiquitin-protease promoting degrada-tion of JEV NS3 was not dependent on C19orf66. These findings confirmed that C19orf66 promotes the degradation of the NS3 proteins in JEV through the lysosome pathway. Nonetheless, C19orf66-209 and C19orf66-Zincmut could possess other virus inhibition mechanisms.
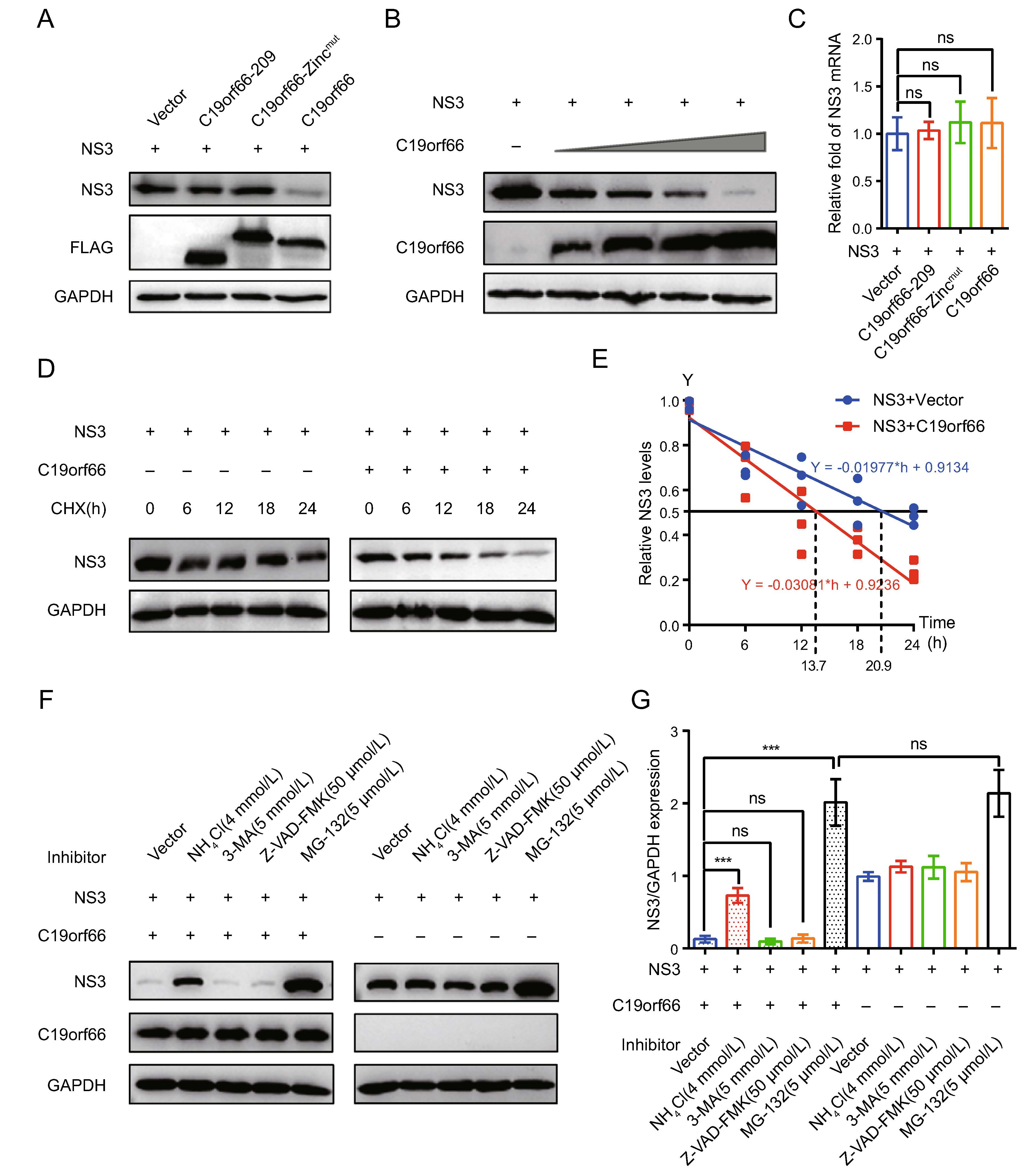
Figure 6. C19orf66 promotes degradation of JEV NS3 protein through lysosome pathway. A 293T cells were co-transfected with NS3 plasmid (0.5 μg) and C19orf66-209 plasmid (0.5 μg), C19orf66-Zincmut plasmid (0.5 μg), C19orf66 plasmid (0.5 μg) or control plasmid (0.5 μg). At 24 h post-transfection, cell lysates were collected for Western blot analysis using FLAG mAb and NS3 mAb. B Western blotting was used to analyze lysates from 293T cells co-transfected with plasmids encoding NS3 (0.7 μg) and increasing amounts of plasmid encoding C19orf66 (0, 0.1, 0.2, 0.4 or 0.8 μg). C Quantitative real time PCR was used to analyze NS3 mRNA abundance in 293T cells co-transfected with NS3 plasmid (0.6 μg) and with C19orf66-209 plasmid (0.4 μg), C19orf66-Zincmut plasmid (0.4 μg), C19orf66 plasmid (0.4 μg) or control plasmid (0.4 μg) for 24 h. D, E 293T cells co-transfected with plasmids encoding NS3 (0.6 μg) and C19orf66 (1 μg) or empty vector (1 μg) were treated with cycloheximide (100 μg/mL) for 0, 6, 12, 18 or 24 h. The stability of NS3 was detected via Western blot analysis. The protein levels of NS3 or C19orf66 were normalized to the level of GAPDH using band intensity values. The half-life (t1/2) of NS3 in 293T cells co-transfected with C19orf66 was determined through linear regression analysis. F, G 293T cells co-transfected with NS3 plasmid (0.3 μg) and C19orf66 plasmid (0.7 μg) or vector plasmid (0.7 μg) were treated with NH4Cl (4 mmol/ L), 3-MA (5 mmol/L), Z-VAD-FMK (50 lmol/L) or MG-132 (5 lmol/L) for 12 h. The expression levels NS3 protein in cells was detected through Western blot analysis. The gray value of NS3 and GAPDH was analyzed using ImageJ and the ratios of NS3 to GAPDH were calculated. The data were expressed as the means ± SDs from three repeat experiments using t-test. ***P < 0.001.
C19orf66 Expression Is Dose-Related to IFN-α and Is Up-Regulated by JEV Infection
Overexpression of C19orf66 in 293T Cells Inhibited JEV Replication
C19orf66 Silencing in HeLa and A549 Cells Resulted in Increased JEV Replication
C19orf66 Down-regulates the Ratio of NS1' to NS1 in JEV Infected Cells
The Zinc-Finger Motif and 164–199 Amino Acids of C19orf66 are Crucial for Its Inhibition Function on JEV -1 PRF
C19orf66 Down-regulates the Expression of JEV NS3 Protein
-
ISG is considered to be an important factor in the antiviral immune response against JEV. Herein, C19orf66 was found to inhibit JEV proliferation in different ways as a host factor. It specifically inhibited the -1 PRF of JEV, leading to decreased NS1' protein production. Amino acids 164–199 and the Zinc-finger domain of C19orf66 were indispensable in inhibiting JEV -1 PRF. In addition, C19orf66 down-regulated the expression of NS3 protein in 293T cells via the lysosome pathway.
Programmed -1 ribosomal frameshifting is a universal translation recoding mechanism present in various prokaryotes and eukaryotes (Caliskan et al. 2015; Atkins et al. 2016). The HIV Gag to frameshift-derived Gag-Pol polyprotein ratio is important for HIV propagation. Both HIV and JEV utilize a programmed -1 ribosomal frameshifting because they have a slippery heptanucleotide sequence and a 3' structural RNA frameshift stimulator.
C19orf66 plays an important role in regulating the expression of JEV NS1' protein by inhibiting -1 PRF. It has a significant frameshift inhibitory effect in the early stages of viral infection, as evidenced by the ratio of NS1' to NS1. However, when pNS1-NS2A and pC19orf66 are co-trans-fected into 293T cells, NS1' to NS1 ratio is significantly reduced. The reduction is similar in three JEV strains: NJ2008, HN2007 and SA14-14-2A66G. The uniqueness of NS1' expression in the neurogenic Flavivirus of the Japanese encephalitis serogroup has sparked significant interest in NS1' functions related to viral virulence. Although the specific NS1' functions remain unknown, several studies have investigated the potential roles of the NS1' protein. The NS1' of JEV antagonizes host MAVS by regulating miR-22, thereby inhibiting IFN-I production while promoting virus replication (Zhou et al. 2020).
Kinast et al. (2020) found that the expression of C19orf66 can change the formation of HCV-induced membrane net-work and weaken the increase of phosphatidylinositol-4-phosphate (PI4P) induced by HCV. They also found that C19orf66 and C19orf66-209 act on HCV replicons restrict HCV replication from different aspects. Herein, C19orf66-Zincmut and C19orf66-209 were employed to analyze the functional domain of C19orf66 because it exists in two iso-forms under natural conditions and both restricted JEV reproduction variably. The Zinc-finger domain and 164 to 199 amino acids of C19orf66 were essential in inhibiting -1 PRF of JEV. Nonetheless, the unknown virus restriction capacity of C19orf66-209 and C19orf66-Zincmut implies that C19orf66 restricts JEV via diverse mechanisms, which can be focused on limiting the spread of the virus.
C19orf66 down-regulated the expression of NS3 protein in 293T cells in a lysosome-dependent manner. This find-ing is consistent with the recent report that C19orf66 can interact with ZIKV NS3, shorten the half-life of NS3, and degrade NS3 via the lysosome-dependent pathway, thereby reducing virus replication (Wu et al. 2020). Notably, the JEV NS3 protein is primarily degraded via the ubiquitin– proteasome pathway in non-overexpressed C19orf66, whereas ZIKV NS3 protein is not affected by the ubiqui-tin–proteasome. NH4Cl is a weak base known to inhibit lysosomal hydrolases by reducing the acidification of the lysosomal compartment (Makki et al. 2008; Saftig and Klumperman 2009; Mizushima et al. 2010). Herein, NH4Cl significantly reversed JEV NS3 protein degradation medi-ated by C19orf66, indicating that C19orf66-mediated degradation of the JEV NS3 protein is dependent on lysosomal pH. Future studies should thus explore the interaction mechanisms of lysosome-related molecules with C19orf66 in the degradation of the JEV NS3 protein.
Previous studies postulate that C19orf66 is localized to ribonucleoprotein particles (RNP, P body) of uninfected cells, which binds to DENV protein to form viral replica-tion complexes. Moreover, C19orf66 is associated with various RNA decay-associated proteins such as HuR, MOV10, UPF1, LARP1 and PABPC1. C19orf66 interacts with viral RNA to provide binding specificity for DENV RNA through C19orf66-PABPC1-LARP1 complex, thereby interfering with normal viral replication (Suzuki et al. 2016; Balinsky et al. 2017).
We speculate that C19orf66-209 and C19orf66-Zincmut do not target JEV NS1' and NS3 proteins to inhibit the virus. From the amino acid sequence of C19orf66-209 and C19orf66-Zincmut, the domains that bind to certain host factors are retained, indicating that they may also interact with host factors to regulate the virus replication cycle. In the same line, the study puts forward three hypotheses regarding the inhibition mechanisms of C19orf66-209 and C19orf66-Zincmut: Hypothesis 1, the host factors, PAPBC1 and LARP1, form replication complexes with C19orf66-209 or C19orf66-Zincmut, which hinder the circularization of RNA during JEV replication; Hypothesis 2, the host factors MOV10, UPF1, and HuR co-localize with C19orf66-209 or C19orf66-Zincmut, thereby inducing the decay of JEV RNA; Hypothesis 3, C19orf66-209 interacts with the non-structural proteins of JEV, thereby inhibiting the PI4KA activation process and destroying the non-structural protein functions, causing the host's cells to form viral replication organelles. These hypotheses give a direction for our future research.
C19orf66, a novel interferon-stimulated gene product, inhibits JEV replication through different pathways. The findings of this study provide baseline information for future development of broad-spectrum antiviral agents against JEV.
-
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 31772756), the National Key Research and Development Program of China (Grant No. 2016YFD0500402) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.
-
RC and DY designed the study, interpreted the data and drafted the paper; DY and YZ performed most of experiments described in the manuscript; JP and XY performed part of the experiment and helped a lot with the experiments; DY and YZ analyzed the data; YZ, ZL, SX and RC provided critical suggestions of the manuscript. All authors read and approved the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.


 DownLoad:
DownLoad: