














-
Dear Editor,
It has been more than 1 year since China reported the first case of African swine fever (ASF) infection in August 2018, and the epidemic situation remains severe (China News Service 2019). According to reports from the Ministry of Agriculture and Rural Affairs, China has reported 160 cases of ASF, which resulted in nearly 1.2 million pigs being killed, as of November 21, 2019 (China News Service 2019). ASF is an acute febrile, hemorrhagic and fulminating infectious disease, and would reach 100% case fatality rate to pigs (Gallardo et al. 2015). The causative pathogen, African swine fever virus (ASFV), is a doublestranded DNA virus with a genome of 170–193 kb belonging to the Asfarviridae family (Galindo and Alonso 2017; Gallardo et al. 2015). A recent study has revealed that ASFV maintains a core genome of 102 ORFs and has 168 dispensable genes (Wang et al. 2019). Thus, the complexed genomic features of ASFV require more attentions. By using the next generation sequencing (NGS) and the single molecule real-time sequencing (SMRT-seq), a couple of Chinese ASFV genomes have been uncovered (Bao et al. 2019; Wen et al. 2019; Jia et al. 2019). Compared to NGS, SMRT-seq has the advantage of long read length and can generate sequencing data containing the original single base modification information, which can be identified through the state-of-art bioinformatic procedures (Senol Cali et al. 2019; Simpson et al. 2017). DNA methylation is a chemical modification common in animal and plant genomes. It refers to the catalytic transfer of methyl groups on active methyl compounds (such as s-adenosine methionine) to other compounds under the catalysis of DNA methyltransferase (DNMT), mainly forming 5-methylcytosine (5-mC), 6-methyladenine (6-mA), 5-hydroxymethylcytosine (5-hmC), etc. DNA methylation, which triggers the epigenetic regulatory mechanism, has been proved to play important roles in gene expression and regulation, embryonic development, and disease-related aspects (Gouil and Keniry 2019). Whether ASFV genome has DNA methylation and epigenetic regulation is to be discerned.
In a previous study, we have sequenced an endemic strain and obtained a complete genome ASFV/pig/China/CAS19-01/2019 (accession number: MN172368, BioSample of Genome Sequence Archive: SAMC072713) by using Nanopore sequencing technique (Jia et al. 2019). CAS19-01 is an ASFV genotype II strain isolated from a clinical tissue sample of a sick pig in Zhuhai. Tissue DNA was extracted and sequenced on Nanopore's promethION platform. Once 100 Gb data was generated, sequencing was terminated and only reads with a quality score > 7 were screened (Fig. 1A). We previously obtained 8, 517 virus reads in fastq format by mapping to the ASFV/HLJ-18 (accession number: MK333180) genome using BWA v0.7.15 (Wen et al. 2019; Li and Durbin 2009), and here we used the tool fast5seek to trace the source to find their corresponding original fast5 files. In order to screen the potential methylated nucleotides, we applied the software suite Tombo, which is a tool set for analyzing and visualizing modified nucleotides from nanopore sequencing data (Stoiber et al. 2017). We implemented the alternative model of Tombo to detect the m5C and m6A modifications in the CAS19-01 genome and output the corresponding scores. The higher the score, the more likely the modification will occur. The results showed that 99% of the scores are between 0 and 0.90, and the predicted sites are evenly distributed along the genome without regional preference (Fig. 1B). Sites with low scores are more susceptible to bias and may be false positives, so we discarded the outputs which scored below 0.9 and obtained 500 m5C and 1340 m6A modifications (Fig. 1C). These potential sites did not show significant strand specificity, but it was unexpected that the number of m6A was much more than m5C. Next, we examined the base composition near the m5C and m6A modification sites of the score top2 (Fig. 1D, 1E). Tombo is a testing-based detection pipeline, which simplifies the comparison of the raw signal level between the sample to be tested and the alternative model into a statistical problem, and obtains statistically significant P value through the two-step test of Mann–Whitney U-test and Fisher's test to predict methylation modification (Stoiber et al. 2017). The results showed that in the detection of m5C, there is a significant difference in the position 141, 427 of the negative chain and the position 51, 922 of the positive chain of CAS19-01 compared with model (in black) (Fig. 1D). Similarly, in the detection of m6A, adenine at position 99, 786 on the negative chain and position 51, 302 on the positive chain are highly likely to be modified (Fig. 1E).
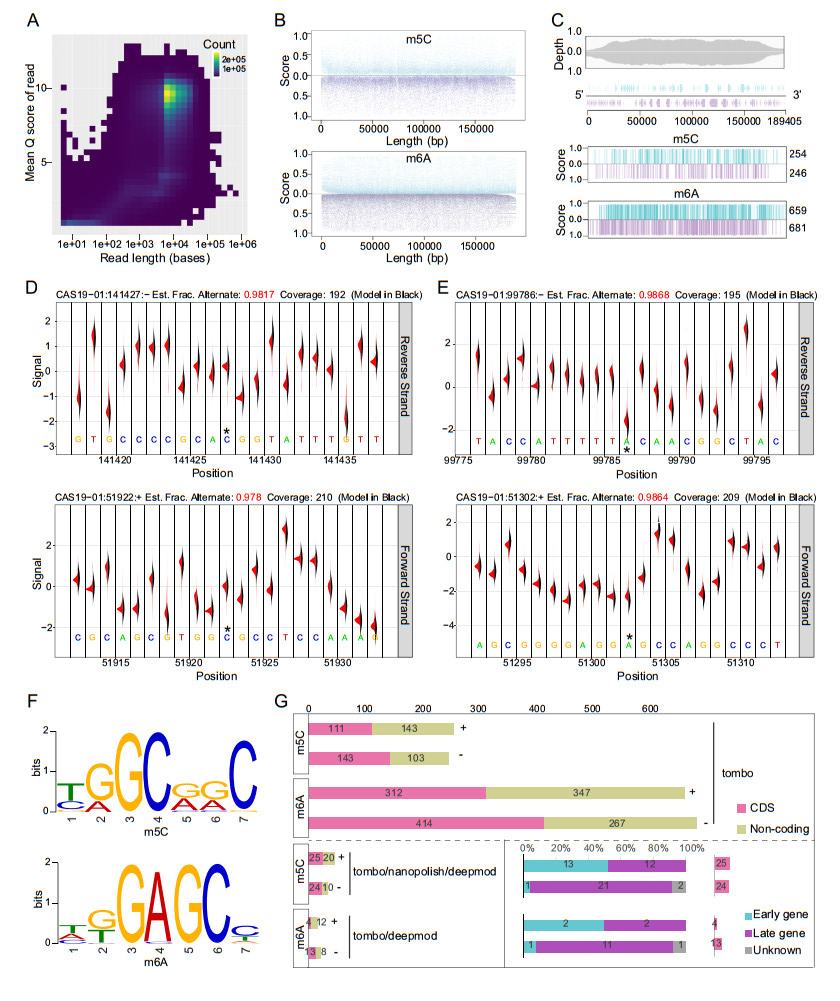
Figure 1. Detection of DNA methylations in the genome of African swine fever virus strain CAS19-01 by nanopore sequencing. The raw fast5 data generated by nanopore sequencing were used to detect electrical signals to determine the presence of modifications on the DNA. A Correlation plot between the quality score and length of each read generated by promethION was shown, and only reads with a quality score >7 were used in this study. B The distribution of m5C and m6A sites predicted by Tombo along the ASFV genome and the corresponding score values. Blue represents the forward strand and purple represents the reverse strand. C The sequencing depth and coverage along the CAS19-01 genome were shown, and the predicted methylation sites with Tombo score >0.9 were left after further screened based on B. D, E The base composition and signal value near the site most likely to be modified by 5-methylcytosine and 6-methyladenine predicted by Tombo are shown, respectively. F Motif patterns of 3 nt upstream and downstream of m5C and m6A sites in CAS19-01, respectively. G Comparison for nanopore data and regional distribution of predicted sites from three methylation detection tools. In terms of the intersection of predicted results, modifications on the reverse strand often occur in the coding region of late genes.
To further explore the special patterns of these two types of modification in ASFV, we extracted 100 genome sequences surrounding unique genomic positions which with the largest estimated fraction of modified bases, and used MEME Version 5.1.0 (Bailey and Elkan 1994) to find motifs (Fig. 1F). We speculated that methylation modification may affect transcriptional regulation, so we searched JASPAR2020 website to see if these motifs might be potential transcription factor binding sites, and the results did confirm our conjecture that the functions of transcription factors highly related to these motif are mainly focus on transcription regulation, DNA replication and differentiation (Supplement Table S1). In addition, we used other two tools, nanopolish (Simpson et al. 2017) and deepmod (Liu et al. 2019), to detect methylation, and listed the sites information that matched the prediction of Tombo in the results (Supplement Table S1). The number of potential methylation sites in coding DNA sequence (CDS) region and non-coding region is not significantly different, so we further investigated which viral genes the methylation sites were distributed on, and found that m5C and m6A modifications on the negative strand were concentrated on the late genes (Fig. 1G, Supplement Table S1) (Cackett et al. 2019).
There are mixed opinions about whether there is a methylation modification in the ASFV genome. Previous studies on the BA71V strain showed methylation at its 5' cap (Salas et al. 1981), while a study last year showed that there is no methylation within the genome but the possibility of modification is not ruled out (Weber et al. 2018). Studies have reported that the methylation of virus-specific genes appears to be involved in the transition from lytic infection to latent infection, and that cytoplasmic virus DNA appears to be consistently methylated (Hoelzer et al. 2008). Our hypothesis is that ASFV, as a large cytoplasmic virus, may try to strengthen its own DNA replication through epigenetic modification after infecting host cells, and correspondingly, the host will invoke some mechanisms to prevent its proliferation, and methylation may be one of the ways. Here in our study testing-based and model-based methods both revealed the possible methylation modification within the genome of the endemic genotype II ASFV strain CAS19-01. At present, it is believed that m5C modification mainly plays a role in inhibiting gene expression, while m6A increasingly shows the role of activating some genes (Gokhale et al. 2020; Hoelzer et al. 2008), and our results showed that these two modifications exist simultaneously in the CAS19-01 genome (Fig. 1). It is speculated that may be the result of checks and balances between the virus and the host, which is likely to be achieved by inhibiting or enhancing the binding of transcription factors, but the specific mechanism is still unclear. In addition, there are complex cell-types in many different stages of infection in the infected tissue, which may lead to a mix of multiple methylation patterns and thus affect the accuracy of experimental results. Therefore, in follow-up studies, not only more epidemic strains need to be collected from multiple regions, but also experiments at the level of single type cell-culture need to be done to describe a more complete and accurate methylation profile of ASFV, which is essential for understanding virus-host interactions.
In summary, we explored the potential m5C and m6A methylation modifications of the genotype II ASFV genome using an unsupervised learning method, providing new insights into virus-host interactions from the epigenetic level and also laid the foundation for the subsequent work on epigenetics mapping of ASFV.
HTML
-
This work was supported by the Research Project of African Swine Fever of Chinese Academy of Sciences (KJZD-SWL06), the National Natural Science Foundation of China (31941015), the National Key R&D Program of China (2016YFC1200800 & 2018YFC0840402), the China Mega-Project for Infectious Disease (2017ZX10103005-005), the State Key Laboratory of Veterinary Biotechnology Research Fund (SKLVBF20 1902). J.L. is supported by Youth Innovation Promotion Association of CAS (2019091). We acknowledge Lei Zhang (the Center for Instrumental Analysis and Metrology in the Wuhan Institute of Virology, CAS) for supporting in the genome sequencing.
-
The authors declare that they have no conflict of interest.
-
All institutional and national guidelines for the care and use of laboratory animals were followed.

 DownLoad:
DownLoad: